


引言
随着化石资源的枯竭与环境污染问题的日益严重,人们迫切需要寻找可再生资源以替代传统的化石燃料.生物质因其可再生、储量丰富等优势引起了广大研究者的关注.生物质资源主要有两大类:木质纤维素生物质和海洋生物质.其中,纤维素是储量最大的生物质资源,是一种由D-葡萄糖单元通过β-1,4-糖苷键连接组成的有机聚合物.纤维素可水解产生D-葡萄糖,随后D-葡萄糖可通过生物催化或化学催化转化为各种平台化学品,如D-果糖、山梨醇、5-羟甲基糠醛(5-HMF)以及乙酰丙酸等[1
上述两大类生物质对应的单糖均为多羟基化合物,且每种糖分子均有多种异构体.这些异构体的比例受温度、pH值、催化剂种类等多种因素的影响[6].醛糖各构型结构如图1(a)和图1(b)所示[12],对于每种单糖,线性形式是焓最高的异构体,其次是两种呋喃糖形式,两种吡喃糖异构体的生成热最低[13],而作为酮糖的D-果糖在溶液状态下通常呈现出更多的异构体形式(图1(c))[14].单糖的构型分布因其与生物质转化机制密切相关而引起了研究者的极大兴趣,不同的单糖异构体具有不同的反应性[12,14-
图1
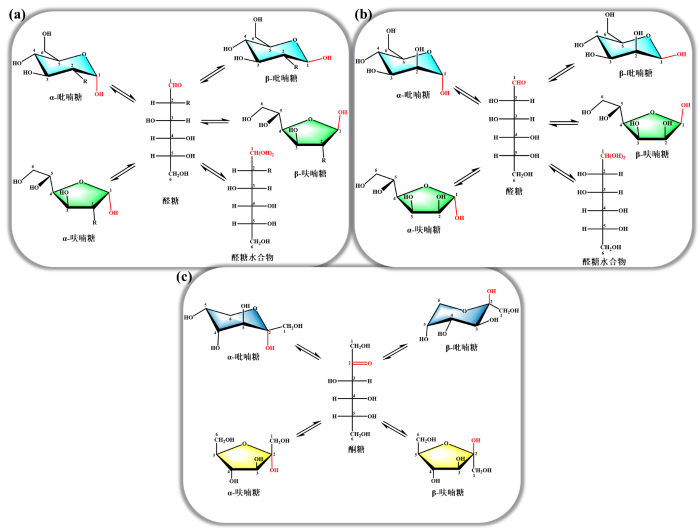
图1
(a)醛糖的异构体结构(R = OH为D-葡萄糖、R = NH2·HCl为D-氨基葡萄糖盐酸盐、R = NHCOCH3为N-乙酰-D-氨基葡萄糖);(b) D-甘露糖的异构体结构;(c) D-果糖的异构体结构
Fig. 1
(a) Tautomeric structure of aldoses (R = OH is D-glucose, R = NH2·HCl is D-glucosamine hydrochloride, R = NHCOCH3 is N-acetyl-D-glucosamine); (b) Tautomeric structure of D-mannose; (c) Tautomeric structure of D-fructose
离子液体(ILs)是常见的绿色溶剂,与传统有机溶剂相比,具有低毒性、低挥发性、高热稳定性和宽液相线范围等优势,其是由有机阳离子与有机或无机阴离子通过强离子键结合形成的化合物,在100 ℃或以下以液体形式存在[21].ILs具有独特的可调特性,使其能够针对各种应用进行定制[22,23].生物质通常不溶于水和一般的有机溶剂,阻碍了生物质的高效利用与转化[2].ILs在生物质溶解、预处理和转化等方面展现出优异性能[2].例如,Hua等[24]合成了Brønsted 酸性离子液体1-(4-磺酸)丁基-3-十六烷基-2-甲基咪唑硫酸氢盐并将其应用于D-木糖脱水制糠醛,在最优条件下,D-木糖转化率为95.3%,糠醛收率为67.5%;Zhao等[25]通过在阳离子中引入Brønsted酸(-SO3H)和在阴离子中引入Lewis酸(AlCl3),设计并合成了一系列Brønsted-Lewis酸性四咪唑基离子液体,在水 : 1-辛醇 = 1 : 2的两相体系中对D-葡萄糖转化为5-HMF表现出良好的催化性能,D-葡萄糖的转化率和5-HMF的收率分别达到95.4%和71.3%;Jia等[26]将弱碱性离子液体1-乙基-3-甲基咪唑醋酸盐用于催化GlcNH2转化,在100 ℃下反应90分钟后产物(脱氧果糖嗪和果糖嗪)的产率达到37%;Chen等[27]系统研究了甲壳素在一系列离子液体中直接转化为3A5AF的过程,其中在氯化1-丁基-3-甲基咪唑中使用硼酸和盐酸作为添加剂,在180 ℃下反应1 h后产物3A5AF收率为6.2%.上述研究工作阐明了咪唑基离子液体作为反应介质和催化剂用于生物炼制的可行性,但领域内一直缺乏对于糖类小分子在上述离子液体中构型分布规律的系统性研究.
核磁共振(NMR)技术被广泛用于分析各种有机分子结构和分子间相互作用,并可以确定反应中间体结构以及揭示反应途径和机制[28
本文采用NMR技术研究了25 ℃条件下醛糖(D-葡萄糖、D-甘露糖、GlcNAc和D-氨基葡萄糖盐酸盐)与酮糖(D-果糖)在不同结构的咪唑基离子液体中的构型分布规律,发现两性金属氯化物与氟原子的引入可能会导致更高比例的呋喃糖的产生.所考察的咪唑基离子液体包括:酸性离子液体(1-磺酸丁基-3-甲基咪唑硫酸氢盐、1-丁基-3-甲基咪唑氯化锌盐)、弱碱性离子液体(1-丁基-3-甲基咪唑醋酸盐)和含氟功能化离子液体(包括1-丁基-3-甲基咪唑四氟硼酸盐、1-丁基-3-甲基咪唑六氟磷酸盐以及1-丁基-3-甲基咪唑三氟乙酸盐等)等.本研究给出了单糖在咪唑基离子液体中构型分布的基础信息,为未来用于单糖转化的咪唑基离子液体的选择与设计提供了指导,同时有利于进一步了解生物质在咪唑基离子液体中的转化机理.
1 实验部分
1.1 仪器与试剂
1.1.1 实验试剂
1-磺酸丁基-3-甲基咪唑硫酸氢盐([HSO3-BMIM]HSO4,99%)、1-丁基-3-甲基咪唑醋酸盐([BMIM]OAc,98%)、1-丁基-3-甲基咪唑三氟乙酸盐([BMIM]TA,99%)、1-丁基-3-甲基咪唑四氟硼酸盐([BMIM]BF4,99%)和氯化1-丁基-3-甲基咪唑([BMIM]Cl,99%)购自上海成捷化学有限公司;D-果糖(分析纯)购自上海晶纯生化科技有限公司;D-葡萄糖(分析纯)、D-甘露糖(99%)、N-乙酰-D-氨基葡萄糖(GlcNAc,98%)、D-氨基葡萄糖盐酸盐(GlcNH2·HCl,≥99%)、1-(2-羟乙基)-3-甲基氯化咪唑([HOEtMIM]Cl,98%)、1-(3-氰丙基)-3-甲基咪唑氯盐([CPMIM]Cl,≥98%)、1-苄基-3-甲基咪唑氯盐([BzMIM]Cl,98%)和1-烯丙基-3-甲基氯化咪唑([AMIM]Cl,96%)购自阿拉丁试剂公司;氯化锌(98%)、1-丁基-3-甲基咪唑六氟磷酸盐([BMIM]PF6,97%)、1-丁基-3-甲基咪唑双三氟甲磺酰亚胺盐([BMIM]Tf2N,99%)、1-丁基-3-甲基咪唑对甲苯磺酸盐([BMIM]TS,≥98%)、1-丁基-3-甲基咪唑甲磺酸盐([BMIM]MeSO3,99%)和1-丁基-3-甲基咪唑三氟甲磺酸盐([BMIM]OTf,97%)购自麦克林试剂公司;氘代二甲基亚砜(DMSO-d6,99.8 atom% D,0.03%(V/V) TMS)购自安耐吉化学公司.所有试剂在使用前未进行进一步的纯化.离子液体结构如图2所示,其中1-丁基-3-甲基咪唑氯化锌盐([BMIM]ZnCl3)按照1.2节中的方法制备.
图2

1.1.2 实验仪器
Bruker Avance 400 MHz核磁共振波谱仪(配备5 mm PABBO BB/19F-1H/D变温探头)购自德国布鲁克仪器公司;电子天平(AL204)购自梅特勒-托利多仪器有限公司.
1.2 离子液体的制备
图3
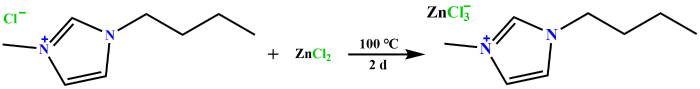
1.3 单糖在离子液体/DMSO-d6中的构型转化平衡过程
称取10 mg离子液体与等摩尔量的单糖置于1.5 mL离心管中,加入500 μL DMSO-d6振荡使其快速溶解,迅速转移至5 mm NMR样品管中,立即进行定量氢谱测试(需要注意的是:由于样品制备时间、NMR测试中锁场与匀场时间等因素的影响,零点的指认存在1~3 min的误差).之后根据单糖的构型变化情况对样品于25 ℃下控温原位监测,确定单糖在离子液体作用下构型转化达到平衡所需要的时间(单糖在12 h以内各构型比例保持不变即认为达到平衡)和平衡时不同单糖异构体的比例(单糖某构型的NMR信号积分面积除以单糖各构型信号积分面积和计算获得).测试样品平衡后的定量碳谱与1H-13C HSQC谱图,以进一步确定样品在离子液体作用下的变化情况和单糖异构体的平衡比例.
1.4 NMR实验
1D和2D NMR实验均在Bruker Avance 400 MHz核磁共振波谱仪(配备5 mm PABBO BB/19F-1H/D变温探头)上完成.
为了保证定量氢谱的定量准确性,需设置D1≥5T1[36].定量氢谱(1H qNMR)的测试参数如下:脉冲序列为zg,弛豫延迟(D1)为20 s,采样次数(NS)为4,谱宽(SWH)为8 012.8 Hz.
1H-13C HSQC的测试参数如下:脉冲序列为hsqcedetgpsisp2.3;采样数据点阵(TD)为t2×t1=2 048×256;采样次数(NS),4;F2(1H)和F1(13C)维的谱宽(SWH)分别为4 795.396 Hz和16 668.479 Hz.
实验所得NMR谱图和数据由Bruker Topspin 3.1和MestReNova 14.0等软件处理.定量氢谱数据处理使用了GSD(Global Spectral Deconvolution)方法,以对重叠谱峰进行拟合,保证其积分面积的准确性.
2 结果与讨论
2.1 D-葡萄糖在咪唑基离子液体/DMSO-d6中的构型分布
如图S1所示,以OH-1作为定量分析信号,当平衡时间延长到12 h时,D-葡萄糖在DMSO-d6中的β-吡喃糖构型比例由20 min时的2.91%增长至18.03%.呋喃糖构型因浓度太低而无法准确定量.D-葡萄糖的β-吡喃糖构型比例在咪唑基离子液体/DMSO-d6中随时间变化的曲线如图4所示.D-葡萄糖最初主要以α-吡喃糖形式存在,随后α-吡喃糖逐渐向β-吡喃糖转化直至达到平衡.如图S2所示,选择各异构体的OH-1信号(α-吡喃糖:δH 6.22;β-吡喃糖:δH 6.59;α-呋喃糖:δH 5.80;β-呋喃糖:δH 5.73)进行异构体比例的相对定量分析,在[BMIM]MeSO3/DMSO-d6中,β-吡喃糖的比例在12 h以内几乎没有发生变化,然而随着平衡时间的延长,α/β-呋喃糖的信号逐渐出现并增强,在12 h时,β-吡喃糖、α-呋喃糖和β-呋喃糖的占比分别为2.72%、0.43%和0.60%,与D-葡萄糖在DMSO-d6中的构型转化情况相比,[BMIM]MeSO3的添加抑制了α-吡喃糖向β-吡喃糖的转化,促进了呋喃糖的生成;同样选择OH-1信号进行定量分析,在引入氟原子形成的[BMIM]OTf/DMSO-d6中(图4和图S3),当平衡时间延长到12 h时,β-吡喃糖的比例从1.96%增长至5.60%,此时β-呋喃糖的占比为0.53%,α-呋喃糖的比例因含量太低而无法准确定量,由此推测氟原子的引入有利于α-吡喃糖向β-吡喃糖的转化而不利于α/β-呋喃糖的形成,[BMIM]OTf的添加减缓了D-葡萄糖的构型转化速度;如图S4所示,选择OH-1信号对α/β-吡喃糖的相对比例进行分析,当平衡时间延长到12 h时,β-吡喃糖在[BMIM]TS/DMSO-d6中的比例由1.96%增长至2.91%,α/β-呋喃糖因信号太弱无法进行定量,上述构型分布结果可能是由于[BMIM]TS中阴离子的负电荷部分离域到苯环上,从而导致亲核性减弱,且苯环分子尺寸较大,可能会阻碍离子液体与单糖分子的作用,[BMIM]TS的添加抑制了D-葡萄糖的构型转化.
图4
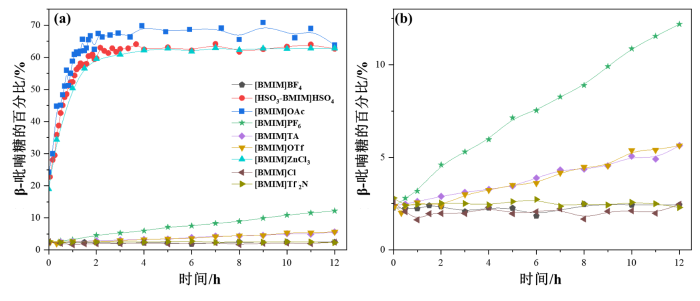
图4
(a) D-葡萄糖的β-吡喃糖构型比例在不同咪唑基离子液体/DMSO-d6中随时间的变化(n(D-葡萄糖) : n(IL) = 1 : 1,25 ℃);(b)局部放大图
Fig. 4
(a) The proportion of β-pyranose of D-glucose over time in imidazolyl ionic liquids/DMSO-d6(n(D-glucose) : n(IL) = 1 : 1, 25 ℃); (b) Partial enlarged figure
以OH-1信号作为定量分析信号,D-葡萄糖在[BMIM]PF6/DMSO-d6中的β-吡喃糖的比例在12 h后由初始时的1.96%增长至12.42%(图4和图S5),此时β-呋喃糖的比例为0.49%,α-呋喃糖无法定量,[BMIM]PF6的添加降低了D-葡萄糖的构型转化速度.如图4和图S6~S8所示,我们同样选择了各异构体的OH-1信号进行分析,在[BMIM]Cl/DMSO-d6、[BMIM]BF4/DMSO-d6以及[BMIM]Tf2N/DMSO-d6中,β-吡喃糖的比例在12 h以内几乎没有发生变化,保持在2%~3%,α/β-呋喃糖无法定量,[BMIM]Cl、[BMIM]BF4和[BMIM]Tf2N的引入抑制了D-葡萄糖的构型转化.如图S9~S12所示,选择OH-1信号进行定量分析,在功能化离子液体[HOEtMIM]Cl/DMSO-d6、[AMIM]Cl/DMSO-d6、[BzMIM]Cl/DMSO-d6和[CPMIM]Cl/DMSO-d6中,β-吡喃糖的比例在平衡时间延长到12 h时几乎没有发生变化,约为2%~3%,其中在[AMIM]Cl中检测到0.10%的α-呋喃糖和0.19%的β-呋喃糖,在[BzMIM]Cl/DMSO-d6中检测到0.19%的α-呋喃糖和0.40%的β-呋喃糖,在[CPMIM]Cl/DMSO-d6中检测到0.10%的α-呋喃糖和0.10%的β-呋喃糖,[HOEtMIM]Cl、[AMIM]Cl、[BzMIM]Cl和[CPMIM]Cl的添加抑制了D-葡萄糖的构型转化.综上所述,[BMIM]MeSO3的引入可能会促进D-葡萄糖的呋喃糖构型的生成,而其它含氟功能化离子液体、咪唑阳离子功能化离子液体和[BMIM]Cl等的添加则可能会抑制D-葡萄糖的构型转化.
酸性离子液体常用于生物质预处理,且在生物质转化过程中具有优异的催化活性.因此考察了D-葡萄糖在酸性离子液体中的构型分布规律,如图4和图S13所示,选择α-吡喃糖的H-6α信号(δH 3.58)、β-吡喃糖的H-6α信号(δH 3.65)、α-呋喃糖的H-4信号(δH 4.06)和β-呋喃糖的H-4信号(δH 3.98)进行定量分析,在Brønsted 酸性离子液体[HSO3-BMIM]HSO4/DMSO-d6中,β-吡喃糖初始占比为22.78%,之后α-吡喃糖的占比逐渐减小而β-吡喃糖的占比逐渐增大,在130 min时构型分布达到平衡,此时β-吡喃糖的比例为62.50%,相对偏差(平衡时刻的构型比例相对于平衡后各点平均值的偏差)为0.40%,以下相对偏差在异构体比例后的括号内给出.有趣的是,平衡后体系中检测到0.29%的α-呋喃糖和0.47%的β-呋喃糖.Brønsted酸性离子液体中氢质子与D-葡萄糖羟基之间的氢键作用可能是D-葡萄糖构型转化的驱动力,[HSO3-BMIM]HSO4的添加极大地缩短了D-葡萄糖构型转化的平衡时间.如图4和图S14所示,选择各异构体的OH-1信号进行定量分析,D-葡萄糖在Lewis酸性离子液体[BMIM]ZnCl3/DMSO-d6中的构型分布情况与在[HSO3-BMIM]HSO4/DMSO-d6中的类似,β-吡喃糖初始时的比例为18.95%,在4 h时构型分布达到平衡,此时β-吡喃糖、α-呋喃糖和β-呋喃糖的比例分别为61.85%(0.59%)、0.11%和0.45%.Lewis酸性离子液体中ZnCl3—可能与D-葡萄糖OH-1和OH-2的氧原子配位促进单糖的构型转化[6],[BMIM]ZnCl3的引入加快了D-葡萄糖的构型转化过程.
碱性离子液体可用于单糖的转化[26],进一步研究了在弱碱性离子液体[BMIM]OAc作用下D-葡萄糖的构型分布情况.如图4和图S15所示,以α-吡喃糖的H-1信号(δH 4.89)、β-吡喃糖的H-4信号(δH 2.92)和α-呋喃糖的H-1信号(δH 5.16)作为定量分析的信号,在[BMIM]OAc/DMSO-d6中,β-吡喃糖初始时的比例为24.15%,在125 min时各种构型之间的转化达到平衡,此时β-吡喃糖的比例为67.11%(0.15%),α-呋喃糖的比例为0.43%,β-呋喃糖的信号由于与其他信号重叠而无法准确定量,[BMIM]OAc的添加缩短了D-葡萄糖构型转化的平衡时间并进一步促进了α-吡喃糖向其它构型的转化;[BMIM]TA与[BMIM]OAc的区别仅在于其阴离子中的氢原子被氟原子取代而不含氢原子,氟原子的电负性较强,故[BMIM]TA中的阴离子较[BMIM]OAc中阴离子的亲核性减弱而亲电性增强,如图4和图S16,选择OH-1信号进行定量分析,D-葡萄糖在[BMIM]TA/DMSO-d6中的构型转化速率较慢,在平衡时间延长到12 h时,β-吡喃糖的比例仅从初始时的2.39%增长至5.55%,α-呋喃糖和β-呋喃糖的比例分别为0.57%和0.88%,由此可知,氟原子的引入降低了离子液体的碱性,不利于D-葡萄糖的α-吡喃糖构型向β-吡喃糖构型的转化而有利于向α/β-呋喃糖的转化,[BMIM]TA的引入减缓了α-吡喃糖构型向β-吡喃糖构型转化的速度.
由上述实验结果可知,D-葡萄糖各异构体的平衡比例在25 ℃下于酸性离子液体[HSO3-BMIM]HSO4和[BMIM]ZnCl3中相当,在弱碱性离子液体[BMIM]OAc中的β-吡喃糖平衡比例略高于酸性离子液体,这可能是由于[BMIM]OAc具有潜在的亲电亲核双活化催化性能,咪唑阳离子的H-2具有较强的酸性而可以提供质子,醋酸根阴离子具有较强的接受质子的能力,与单糖羟基之间可以形成较强的氢键相互作用,[BMIM]OAc中阴离子与阳离子的协同作用共同促进单糖的构型转化[26];而其它含氟功能化离子液体、咪唑阳离子功能化离子液体和[BMIM]Cl等的添加则可能会抑制D-葡萄糖的构型转化;α/β-呋喃糖在[BMIM]TA中的比例较高.
2.2 D-甘露糖在咪唑基离子液体/DMSO-d6中的构型分布
D-甘露糖与D-葡萄糖互为差向异构体,二者的区别仅在于2号位置的羟基取向不同.图1(b)所示为D-甘露糖的异构体结构,我们研究了D-甘露糖于25 ℃下在酸性离子液体[HSO3-BMIM]HSO4和[BMIM]ZnCl3、碱性离子液体[BMIM]OAc以及含氟功能化离子液体[BMIM]BF4和[BMIM]PF6作用下的构型分布规律,D-甘露糖在DMSO-d6和咪唑基离子液体/DMSO-d6中的1H qNMR随时间变化的叠加图如 图S17~S22所示,参考文献[40-
如图S17,选择吡喃糖的OH-1信号和呋喃糖的H-1信号进行定量分析,D-甘露糖在DMSO-d6中初始时α-吡喃糖、β-吡喃糖和α-呋喃糖的比例分别为95.24%、3.81%和0.95%,平衡时间延长到12 h时,α-吡喃糖、β-吡喃糖和α-呋喃糖的比例分别为88.50%、10.62%和0.88%,β-呋喃糖因信号重叠而无法准确定量.如图5(a)和图S18所示,以H-1信号作为定量分析的信号(α-吡喃糖:δH 4.86,β-吡喃糖:δH 4.54),在[HSO3-BMIM]HSO4/DMSO-d6中,D-甘露糖的β-吡喃糖的比例在平衡时间延长到12 h时未发生变化,12 h时β-吡喃糖的比例为11.60%(0.08%),呋喃糖的浓度较低而无法准确定量,[HSO3-BMIM]HSO4的添加加快了D-甘露糖的构型转化过程;如图5(b)和图S19,选择α-吡喃糖的OH-1信号(δH 6.23)、β-吡喃糖的OH-1信号(δH 6.16)、α-呋喃糖的H-1信号(δH 4.96)和β-呋喃糖的H-1信号(δH 4.89)进行定量分析,D-甘露糖在[BMIM]ZnCl3/DMSO-d6中初始时α-吡喃糖、β-吡喃糖、α-呋喃糖和β-呋喃糖的比例分别为91.33%、6.79%、0.60%和1.29%,在30 min时构型分布达到平衡(氢谱定量存在误差,各构型比例存在微小波动),此时α-吡喃糖、β-吡喃糖、α-呋喃糖和β-呋喃糖的比例分别为86.61%(1.36%)、10.34%(5.90%)、0.96%(10.78%)和2.09%(16.04%).可见,D-甘露糖在Lewis酸性离子液体[BMIM]ZnCl3中的呋喃糖比例较[HSO3-BMIM]HSO4高,且[BMIM]ZnCl3的引入缩短了构型转化的平衡时间.
图5

图5
(a) D-甘露糖的β-吡喃糖比例在咪唑基离子液体/DMSO-d6中随时间的变化(n(D-甘露糖) : n(IL) = 1 : 1,25 ℃);(b)异构体比例在[BMIM]ZnCl3/DMSO-d6中随时间的变化(n(D-甘露糖) : n(IL) = 1 : 1,25 ℃)
Fig. 5
(a) The proportion of β-pyranose of D-mannose over time in imidazolyl ionic liquids/DMSO-d6(n(D-mannose) : n(IL) = 1 : 1, 25 ℃); (b) The proportion of tautomer over time in [BMIM]ZnCl3/DMSO-d6(n(D-mannose) : n(IL) = 1 : 1, 25 ℃)
如图6(a)和图S20所示,选择α-吡喃糖的H-1信号(δH 4.86)、β-吡喃糖的H-5信号(δH 2.95)和α-呋喃糖的H-1信号(δH 4.94)进行定量分析,在[BMIM]OAc/DMSO-d6中,α-吡喃糖、β-吡喃糖和α-呋喃糖初始时的比例分别为82.12%、15.26%和2.62%,平衡时间延长到12 h,各异构体比例基本保持不变,由于[BMIM]OAc与D-甘露糖的羟基和水之间存在较强的氢键相互作用,羟基的活泼氢信号与水信号无法分辨,共同形成一个宽峰,进而导致了定量氢谱中的积分面积存在一定误差,鉴于此,我们采用1H-13C HSQC对D-甘露糖在[BMIM]OAc/DMSO-d6中的平衡比例进行了定量分析,选择各异构体的H-1/C-1信号(α-吡喃糖:4.86/94.1,β-吡喃糖:4.48/94.6,α-呋喃糖:4.94/101.8)进行积分,得到α-吡喃糖、β-吡喃糖和α-呋喃糖的平衡比例分别为84.0%、15.1%和0.8%(如图7),β-呋喃糖因信号重叠而无法定量,[BMIM]OAc的添加缩短了构型转化的平衡时间并进一步促进了α-吡喃糖向β-吡喃糖的转化.
图6
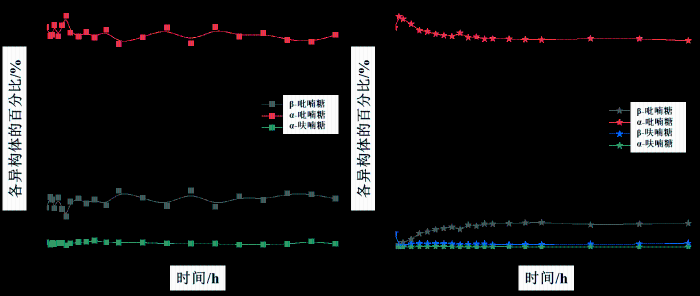
图6
(a) D-甘露糖的异构体比例在[BMIM]OAc/DMSO-d6中随时间的变化(n(D-甘露糖) : n(IL) = 1 : 1,25 ℃);(b) D-甘露糖的异构体比例在[BMIM]PF6/DMSO-d6中随时间的变化(n(D-甘露糖) : n(IL) = 1 : 1,25 ℃)
Fig. 6
(a) The proportion of tautomer of D-mannose over time in [BMIM]OAc/DMSO-d6(n(D-mannose) : n(IL) = 1 : 1, 25 ℃); (b) The proportion of tautomer of D-mannose over time in [BMIM]PF6/DMSO-d6(n(D-mannose) : n(IL) = 1 : 1, 25 ℃)
图7
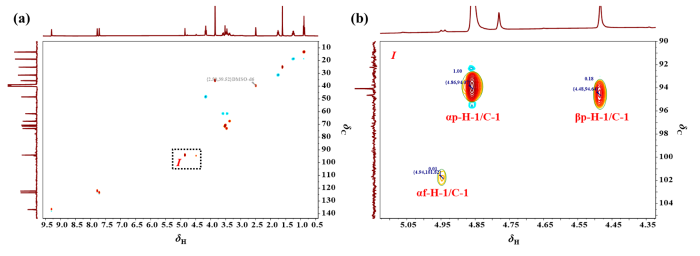
图7
(a) D-甘露糖在[BMIM]OAc/DMSO-d6中平衡后的1H-13C HSQC谱图(n(D-甘露糖) : n(IL) = 1 : 1,25 ℃);(b) 区域Ⅰ的局部放大图
Fig. 7
(a) The 1H-13C HSQC spectra of D-mannose in [BMIM]OAc/DMSO-d6 after equilibrium(n(D-mannose) : n(IL) = 1 : 1, 25 ℃); (b) The partial enlarged figure of area Ⅰ
如图5(a)和图S21,通过OH-1信号分析两种吡喃糖的相对比例,在[BMIM]BF4/DMSO-d6中,β-吡喃糖的比例在12 h以内保持不变,在平衡时间延长到12 h时,β-吡喃糖的比例为0.69%(17.72%),[BMIM]BF4的引入抑制了D-甘露糖的构型转化;如图6(b)和图S22所示,选择OH-1信号分析吡喃糖的比例,选择H-1信号分析呋喃糖的比例,在[BMIM]PF6/DMSO-d6中,初始时呋喃糖的含量较低而无法准确定量,导致初始点各异构体的比例存在较大误差,30 min时α-吡喃糖、β-吡喃糖、α-呋喃糖和β-呋喃糖的比例分别为95.69%、2.00%、0.81%和1.49%,在11 h时构型分布达到平衡,此时α-吡喃糖、β-吡喃糖、α-呋喃糖和β-呋喃糖的比例分别为86.36%(0.04%)、10.49%(0.80%)、0.93%(7.70%)和2.23%(5.95%),[BMIM]PF6的添加可能会促进呋喃糖的生成.
综上所述,D-甘露糖25 ℃时在各离子液体存在条件下主要以α-吡喃糖形式存在,这可能是由于D-甘露糖的OH-2质子与环氧原子形成分子内氢键使其结构稳定,进而导致离子液体难以与D-甘露糖作用,此外,D-甘露糖的β-吡喃糖中存在不利的邻位相互作用,因此会向更稳定的α-吡喃糖转变[41];β-吡喃糖在[BMIM]OAc中的平衡比例略高;D-甘露糖各异构体的平衡比例在[BMIM]ZnCl3和[BMIM]PF6中相当且其中α/β-呋喃糖比例较高;[BMIM]BF4的添加可能会阻碍D-甘露糖的构型转化.此外,与D-葡萄糖相比,D-甘露糖的呋喃糖比例更高,如果D-甘露糖通过与溶剂结合而重新定向远离分子,则氧轨道应向内指向,分子模型表明,这种排列会增加这些轨道与环氧的竖立副轨道之间的排斥相互作用,不利的相互作用模式的引入可能会因向呋喃糖或α-吡喃糖形式的转变而得到缓解[41].
2.3 N-乙酰-D-氨基葡萄糖和D-氨基葡萄糖盐酸盐在咪唑基离子液体/DMSO-d6中的构型分布
进一步研究了含氮单糖GlcNAc和GlcNH2·HCl于25 ℃下在酸性离子液体[HSO3-BMIM]HSO4和[BMIM]ZnCl3、碱性离子液体[BMIM]OAc以及含氟功能化离子液体[BMIM]BF4和[BMIM]PF6作用下的构型分布规律.GlcNAc在DMSO-d6和咪唑基离子液体/DMSO-d6中的1H qNMR随时间变化的叠加图如图S23~S28所示,参考文献[6,10]对GlcNAc的NMR谱峰进行了指认,选择虚线框中的信号峰对GlcNAc异构体比例进行定量分析.据文献报道,在平衡时间延长到800 min时,GlcNAc于25 ℃下在DMSO-d6中的β-吡喃糖比例保持不变,为5.61%[6].
如图S23,以OH-1作为定量分析信号,平衡时间延长到12 h时,GlcNAc在DMSO-d6中的β-吡喃糖比例由初始的4.76%增长至5.66%.如图8(a)和图S24所示,选择GlcNAc的-NH信号(α-吡喃糖:δH 7.69,β-吡喃糖:δH 7.82)进行相对定量分析,在[HSO3-BMIM]HSO4/DMSO-d6中,β-吡喃糖最初的比例为11.21%,在49 min时构型转化达到平衡,此时β-吡喃糖的比例为23.15%(4.20%),[HSO3-BMIM]HSO4的引入促进了α-吡喃糖向β-吡喃糖的转化;如图8(a)和图S25,选择OH-1信号(α-吡喃糖:δH 6.40,β-吡喃糖:δH 6.49)对吡喃糖的比例进行定量分析,在[BMIM]ZnCl3/DMSO-d6中,β-吡喃糖初始时的比例为18.59%,20 min时构型分布达到平衡,此时β-吡喃糖的比例为20.58%(2.79%).由此可知,β-吡喃糖在[HSO3-BMIM]HSO4中的平衡比例略高于在[BMIM]ZnCl3中的平衡比例,推测GlcNAc在[BMIM]ZnCl3中的构型转化速度较[HSO3-BMIM]HSO4快,[BMIM]ZnCl3的添加同样促进了GlcNAc的构型转化.如图8(a)和图S26所示,选择-NH信号(α-吡喃糖:δH 8.11,β-吡喃糖:δH 8.43)进行定量分析,在[BMIM]OAc/DMSO-d6中,β-吡喃糖最初的比例为8.53%,在1.5 h时构型转化达到平衡,此时β-吡喃糖的比例为21.22%(3.16%),GlcNAc在[BMIM]OAc中的构型转化速度较酸性离子液体慢,[BMIM]OAc的引入促进了α-吡喃糖向β-吡喃糖的转化.如图8(a)和图S27~28,同样选择OH-1信号进行定量分析,平衡时间延长到12 h时,GlcNAc在含氟功能化离子液体[BMIM]BF4和[BMIM]PF6中的各异构体比例保持不变,β-吡喃糖的比例约为5%,这与DMSO-d6中β-吡喃糖的比例接近[6].可以发现,在酸性或碱性离子液体作用下,GlcNAc的β-吡喃糖平衡比例较D-葡萄糖低,这可能是由于GlcNAc的N-乙酰基中的羰基氧原子与异头位点上的羟基质子之间形成分子内氢键,进而降低了N-乙酰基的旋转自由度,且与D-葡萄糖中的羟基相比,N-乙酰基的分子尺寸更大,从而阻碍了离子液体与GlcNAc之间的相互作用[43].
图8
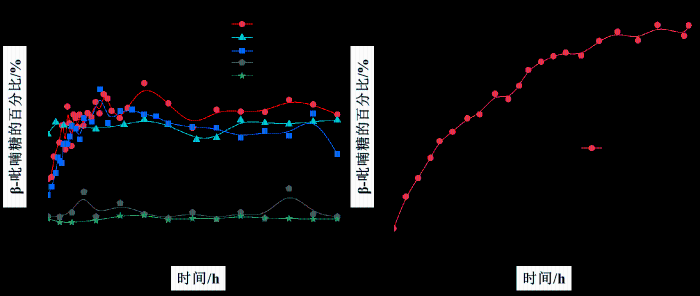
图8
(a) GlcNAc的β-吡喃糖比例在咪唑基离子液体/DMSO-d6中随时间的变化(n(GlcNAc) : n(IL) = 1 : 1,25 ℃);(b) GlcNH2·HCl的β-吡喃糖比例在[HSO3-BMIM]HSO4/DMSO-d6中随时间的变化(n(GlcNH2·HCl) : n(IL) = 1 : 1,25 ℃)
Fig. 8
(a) The proportion of β-pyranose of GlcNAc over time in imidazolyl ionic liquids/DMSO-d6(n(GlcNAc) : n(IL) = 1 : 1, 25 ℃); (b) The proportion of β-pyranose of GlcNH2·HCl over time in [HSO3-BMIM]HSO4/DMSO-d6(n(GlcNH2·HCl) : n(IL) = 1 : 1, 25 ℃)
如图S29所示,选择OH-1信号进行定量分析,GlcNH2·HCl在DMSO-d6中的β-吡喃糖比例在12 h以内保持不变,为2.91%.GlcNH2·HCl在[HSO3-BMIM]HSO4/DMSO-d6中的1H qNMR随时间变化的叠加图如 图S30所示,参考文献[44]对GlcNH2·HCl的NMR信号进行了归属,选择虚线框中的信号峰对GlcNH2·HCl异构体比例进行定量分析.由文献[26]可知,在室温下放置3天后,GlcNH2在DMSO-d6中的构型转化比例极小,可忽略不计.如图8(b)和图S30所示,选择NH2·HCl信号(α-吡喃糖:δH 8.00,β-吡喃糖:δH 8.11)进行相对定量分析,GlcNH2·HCl在[HSO3-BMIM]HSO4/DMSO-d6中的β-吡喃糖比例由最初的2.44%增长为平衡时的28.38%(0.06%),平衡时间为72.83 h,[HSO3-BMIM]HSO4的添加促进了GlcNH2·HCl的构型转化;如图9所示,在[BMIM]ZnCl3/DMSO-d6中,随着平衡时间的延长,GlcNH2·HCl的α-吡喃糖构型逐渐向β-吡喃糖构型转化,且逐渐生成了少量的果糖嗪(FZ).如图S31,GlcNH2·HCl在[BMIM]OAc/DMSO-d6中的β-吡喃糖比例迅速增长且有胡敏素的生成,随着平衡时间的延长,GlcNH2·HCl的吡喃糖信号向低场移动,表明GlcNH2·HCl与[BMIM]OAc之间的氢键作用能够促使GlcNH2·HCl的构型转化与进一步反应.如图S32和33所示,选择OH-1信号(α-吡喃糖:δH 7.17,β-吡喃糖:δH 7.52)进行定量分析,GlcNH2·HCl在[BMIM]BF4/DMSO-d6和[BMIM]PF6/DMSO-d6中的构型分布在12 h以内保持不变,β-吡喃糖的比例约为4%~5%,[BMIM]BF4和[BMIM]PF6的引入可能促进了GlcNH2·HCl的构型转化.
图9
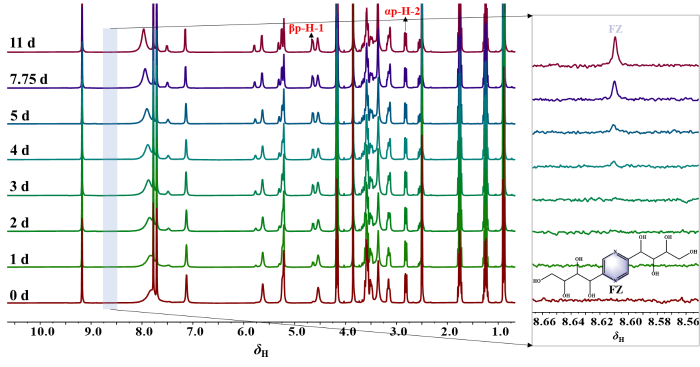
图9
GlcNH2·HCl在[BMIM]ZnCl3/DMSO-d6中的原位1H qNMR随时间变化叠加图(n(GlcNH2·HCl) : n(IL) = 1 : 1,m(IL) = 10 mg,25 ℃)
Fig. 9
The time-progression stack of in situ 1H qNMR of GlcNH2·HCl in [BMIM]ZnCl3/DMSO-d6(n(GlcNH2·HCl) : n(IL) = 1 : 1, m(IL) = 10 mg, 25 ℃)
2.4 D-果糖在咪唑基离子液体/DMSO-d6中的构型分布
如图S35所示,D-果糖在DMSO-d6中初始时β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的比例分别为71.94%、1.44%、19.42%和7.19%,4 h时β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的比例分别为25.91%、4.66%、49.74%和19.69%,8 h时β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的比例分别为24.94%、4.99%、49.38%和20.70%.由此可知,D-果糖在DMSO-d6中构型转化的平衡时间在4~8 h之间.如图S36所示,D-果糖在[HSO3-BMIM]HSO4/DMSO-d6中逐渐反应生成5-HMF,图S37(13C NMR和1H-13C HSQC NMR)进一步证明了5-HMF的结构;如图10(a),在[BMIM]ZnCl3/DMSO-d6中,β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的平衡比例分别为24.27%、5.10%、50.49%和20.15%,平衡时间为10 min,[BMIM]ZnCl3的添加缩短了D-果糖的构型转化平衡时间.
图10
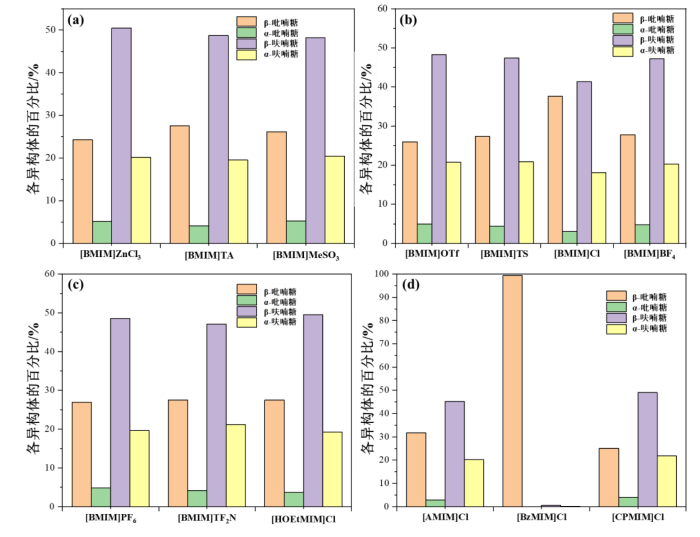
图10
平衡状态下D-果糖在咪唑基离子液体/DMSO-d6中的构型分布(n(D-果糖) : n(IL) = 1 : 1,25 ℃)
Fig. 10
Tautomer distributions of D-fructose at equilibrium in imidazolyl ionic liquids/DMSO-d6(n(D-fructose) : n(IL) = 1 : 1, 25 ℃)
如图S38所示,由于样品NMR信号重叠严重,难以通过1H qNMR对D-果糖在[BMIM]OAc/DMSO-d6中的构型分布情况进行定量,故我们在该样品平衡12 h后对其进行了碳谱测试(如图S39),此时β-吡喃糖、β-呋喃糖和α-呋喃糖的平衡比例分别为28.49%、48.15%和23.36%;如图10(a),在[BMIM]TA/DMSO-d6中,D-果糖在114 h时构型转化达到平衡,此时β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的比例分别为27.55%、4.13%、48.76%和19.56%,[BMIM]TA的引入减缓了D-果糖的构型转化速度.
如图10(a)所示,在[BMIM]MeSO3/DMSO-d6中,β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的平衡比例分别为26.18%、5.24%、48.17%和20.42%,平衡时间为14 h;如图10(b),D-果糖在[BMIM]OTf/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为25.97%、4.94%、48.31%和20.78%,平衡时间为36 h,在[BMIM]TS/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为27.40%、4.38%、47.40%和20.82%,平衡时间为108 h,[BMIM]MeSO3、[BMIM]OTf和[BMIM]TS的添加减缓了D-果糖的构型转化速度.
如图10(b)所示,D-果糖在[BMIM]Cl/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为37.59%、3.01%、41.35%和18.05%,平衡时间为324 h,[BMIM]Cl的引入可能抑制了D-果糖的β-吡喃糖构型向其它构型的转化,在[BMIM]BF4/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为27.78%、4.72%、47.22%和20.28%,平衡时间为128 h,[BMIM]BF4的添加降低了构型转化速度;如图10(c)所示,在[BMIM]PF6/DMSO-d6中,D-果糖在1 h 40 min时构型分布达到平衡,此时β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的比例分别为26.95%、4.85%、48.52%和19.68%,[BMIM]PF6的引入缩短了构型转化的平衡时间,在[BMIM]Tf2N/DMSO-d6中,β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的平衡比例分别为27.55%、4.13%、47.11%和21.21%,平衡时间为198 h,[BMIM]Tf2N的添加减缓了构型转化速度.
如图10(c)所示,在[HOEtMIM]Cl/DMSO-d6中,D-果糖的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为27.52%、3.67%、49.54%和19.27%,平衡时间为300 h,[HOEtMIM]Cl的引入降低了构型转化速度;如图10(d),D-果糖在[AMIM]Cl/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为31.73%、2.88%、45.19%和20.19%(126 h),在[BzMIM]Cl/DMSO-d6中,当平衡时间延长到12 h时,D-果糖的β-吡喃糖、β-呋喃糖和α-呋喃糖比例分别为99.28%、0.58%和0.15%,[AMIM]Cl和[BzMIM]Cl可能抑制了D-果糖的构型转化,在[CPMIM]Cl/DMSO-d6中的β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖平衡比例分别为25.06%、4.01%、49.12%和21.80%,平衡时间为84 h,[CPMIM]Cl的添加减缓了构型转化速度.
综上所述,D-果糖在[HSO3-BMIM]HSO4中的反应性较强;此外,D-果糖各异构体在[BMIM]ZnCl3、[BMIM]TA、[BMIM]MeSO3、[BMIM]OTf、[BMIM]TS、[BMIM]BF4、[BMIM]PF6、[BMIM]Tf2N、[HOEtMIM]Cl和[CPMIM]Cl中具有相似的平衡比例,构型转化速率由快到慢为:[BMIM]ZnCl3 > [BMIM]PF6 > [BMIM]MeSO3 > [BMIM]OTf > [CPMIM]Cl > [BMIM]TS ≥ [BMIM]TA > [BMIM]BF4 > [BMIM]Tf2N > [HOEtMIM]Cl,在弱碱性离子液体[BMIM]OAc中平衡12 h后的构型分布与上述离子液体中的相近;D-果糖在[BMIM]Cl中的呋喃糖比例较低且构型转化速度较慢;D-果糖在[AMIM]Cl中的呋喃糖比例略低,在[BzMIM]Cl中12 h时呋喃糖的比例仍然是可以忽略的;[BMIM]PF6的引入缩短了D-果糖构型转化的平衡时间,而其它含氟功能化离子液体、咪唑阳离子功能化离子液体和[BMIM]Cl等的添加则可能会抑制D-果糖的构型转化.据文献中报道,D-果糖(0.5 mol/L)25 ℃时在DMSO-d6中构型转化达到平衡需要7.5周,β-吡喃糖、α-吡喃糖、β-呋喃糖和α-呋喃糖的平衡比例分别为27.0%、4.8%、48.1%和20.1%[45],本文研究D-果糖在DMSO-d6作用下的构型转化过程中所用的D-果糖浓度约为0.09 mol/L,平衡时间在4~8 h之间,表明D-果糖浓度会影响其构型转化速度.D-果糖的呋喃糖结构趋向于生成5-HMF,吡喃糖结构趋向于生成副产物,由此可以推断呋喃糖比例高的离子液体体系可能得到更高产率和选择性的5-HMF.
2.5 不同单糖的构型分布规律
D-葡萄糖最初主要以α-吡喃糖形式存在,在酸性离子液体[HSO3-BMIM]HSO4和[BMIM]ZnCl3以及弱碱性离子液体[BMIM]OAc作用下,D-葡萄糖构型转化平衡后主要以β-吡喃糖形式存在且在[BMIM]OAc中的β-吡喃糖比例略高于其他离子液体;D-甘露糖即使在酸性和碱性离子液体作用下仍主要以α-吡喃糖形式存在,然而,相较于D-葡萄糖,D-甘露糖的呋喃糖比例略高,这可能是由于D-甘露糖中不利相互作用模式的引入可能会因向呋喃糖或α-吡喃糖形式的转变而得到缓解;含氮单糖GlcNAc平衡后主要以α-吡喃糖形式存在,GlcNH2·HCl在[BMIM]ZnCl3中可以观察到果糖嗪的生成,在[BMIM]OAc中生成副产物胡敏素;作为酮糖的D-果糖反应性较强,其在[HSO3-BMIM]HSO4中最终几乎完全转化为5-HMF,在其他离子液体中平衡后呋喃糖的比例甚至高于吡喃糖的比例.
在酸性离子液体中,β-吡喃糖的平衡比例由大到小为:D-葡萄糖 > GlcNH2·HCl> GlcNAc> D-甘露糖.说明了单糖取代基的取向和种类对单糖的构型转化具有显著影响.且由实验结果可以推测,两性金属氯化物与氟原子的引入可能会导致更高比例的呋喃糖的产生,例如,D-葡萄糖在[BMIM]TA中的呋喃糖比例较其它离子液体中的略高,D-甘露糖在[BMIM]ZnCl3和[BMIM]PF6中的呋喃糖比例较其它离子液体略高.而其它含氟功能化离子液体、咪唑阳离子功能化离子液体和[BMIM]Cl等的添加则可能会阻碍单糖的构型转化.
由于当前测试条件的局限,在后续工作中可进一步深入探究温度、单糖浓度、离子液体性质等方面对单糖构型分布的影响,进一步借助高场液体NMR及高级NMR方法完成对单糖低浓度构型的准确定量,在分子层面进一步深入解析离子液体对单糖构型分布的影响。
3 结论
本文通过NMR方法研究了25 ℃时醛糖(D-葡萄糖、D-甘露糖、GlcNAc和GlcNH2·HCl)以及酮糖(D-果糖)在不同咪唑基离子液体作用下的构型分布规律.在酸性离子液体中,D-葡萄糖具有更高的β-吡喃糖比例,GlcNH2·HCl次之,D-甘露糖的β-吡喃糖比例最低,GlcNAc的β-吡喃糖比例低于GlcNH2·HCl,说明了单糖取代基的取向和种类对单糖的构型转化具有显著影响.D-葡萄糖和D-甘露糖在碱性离子液体[BMIM]OAc中的β-吡喃糖比例略高于其他离子液体.GlcNH2·HCl在[BMIM]ZnCl3中可以观察到果糖嗪的生成,在[BMIM]OAc中生成副产物胡敏素.D-果糖在[HSO3-BMIM]HSO4中最终几乎完全转化为5-HMF,在其他离子液体中平衡后呋喃糖的比例甚至高于吡喃糖的比例.且由实验结果可以推测,两性金属氯化物与氟原子的引入可能会导致更高比例的呋喃糖的产生.而其它含氟功能化离子液体、咪唑阳离子功能化离子液体和[BMIM]Cl等的添加则可能会抑制单糖的构型转化.本工作为未来用于生物质转化的离子液体的选择与设计提供了指导,同时为生物质转化机理的进一步研究提供了基础数据.
附件材料
附件材料(可在《波谱学杂志》期刊官网
图S1 D-葡萄糖在DMSO-
图S2 D-葡萄糖在[BMIM]MeSO3/DMSO-
图S3 D-葡萄糖在[BMIM]OTf/DMSO-
图S4 D-葡萄糖在[BMIM]TS/DMSO-
图S5 D-葡萄糖在[BMIM]PF6/DMSO-
图S6 D-葡萄糖在[BMIM]Cl/DMSO-
图S7 D-葡萄糖在[BMIM]BF4/DMSO-
图S8 D-葡萄糖在[BMIM]Tf2N/DMSO-
图S9 D-葡萄糖在[HOEtMIM]Cl/DMSO-
图S10 D-葡萄糖在[AMIM]Cl/DMSO-
图S11 D-葡萄糖在[BzMIM]Cl/DMSO-
图S12 D-葡萄糖在[CPMIM]Cl/DMSO-
图S13 D-葡萄糖在[HSO3-BMIM]HSO4/DMSO-
图S14 D-葡萄糖在[BMIM]ZnCl3/DMSO-
图S15 D-葡萄糖在[BMIM]OAc/DMSO-
图S16 D-葡萄糖在[BMIM]TA/DMSO-
图S17 D-甘露糖在DMSO-
图S18 D-甘露糖在[HSO3-BMIM]HSO4/DMSO-
图S19 D-甘露糖在[BMIM]ZnCl3/DMSO-
图S20 D-甘露糖在[BMIM]OAc/DMSO-
图S21 D-甘露糖在[BMIM]BF4/DMSO-
图S22 D-甘露糖在[BMIM]PF6/DMSO-
图S23 GlcNAc在DMSO-
图S24 GlcNAc在[HSO3-BMIM]HSO4/DMSO-
图S25 GlcNAc在[BMIM]ZnCl3/DMSO-
图S26 GlcNAc在[BMIM]OAc/DMSO-
图S27 GlcNAc在[BMIM]BF4/DMSO-
图S28 GlcNAc在[BMIM]PF6/DMSO-
图S29 GlcNH2·HCl在DMSO-
图S30 GlcNH2·HCl在[HSO3-BMIM]HSO4/DMSO-
图S31 GlcNH2·HCl在[BMIM]OAc/DMSO-
图S32 GlcNH2·HCl在[BMIM]BF4/DMSO-
图S33 GlcNH2·HCl在[BMIM]PF6/DMSO-
图S34 D-果糖在[BMIM]BF4/DMSO-
图S35 D-果糖在DMSO-
图S36 D-果糖在[HSO3-BMIM]HSO4/DMSO-
图S37 D-果糖在[HSO3-BMIM]HSO4/DMSO-
图S38 D-果糖在[BMIM]OAc/DMSO-
图S39 D-果糖在[BMIM]OAc/DMSO-
利益冲突
无
参考文献
Efficient preparation of 5-hydroxymethylfurfural from cellulose via one-step combination of mechanical and chemical pre-treatment
[J].DOI:10.1016/j.renene.2024.120748 URL [本文引用: 1]
Imidazolium-based ionic liquids as cellulose solvents: Mechanism and molecular insights
[J].DOI:10.1016/j.biombioe.2025.107758 URL [本文引用: 2]
Catalytic conversion network for lignocellulosic biomass valorization: a panoramic view
[J].The catalytic conversion networks for lignocellulose valorization including reaction routes, reaction types and key steps are comprehensively reviewed. The issues that need to be addressed for large-scale application are proposed.
Efficient conversion of cellulose to lactic acid over yttrium modified siliceous Beta zeolites
[J].DOI:10.1016/j.apcata.2021.118133 URL [本文引用: 1]
Valorization of chitin derived N-acetyl-D-glucosamine into high valuable N-containing 3-acetamido-5-acetylfuran using pyridinium-based ionic liquids
[J].DOI:10.1016/j.molliq.2021.115667 URL [本文引用: 2]
Tautomer distributions of N-acetyl-D-glucosamine in the condition of commonly utilized solvents and catalysts for biorefinery: NMR study
[J].DOI:10.1016/j.molstruc.2021.131995 URL [本文引用: 8]
Non-catalytic synthesis of Chromogen I and III from N-acetyl-D-glucosamine in high-temperature water
[J].DOI:10.1039/c3gc41161c URL [本文引用: 1]
Efficient one-pot synthesis of deoxyfructosazine and fructosazine from D-glucosamine hydrochloride using a basic ionic liquid as a dual solvent-catalyst
[J].DOI:10.1039/C4RA06832G URL [本文引用: 1]
Preparation of 3-acetamido-5-acetylfuran from N-acetylglucosamine and chitin using biobased deep eutectic solvents as catalysts
[J].
DOI:10.1039/D2RE00118G
URL
[本文引用: 1]

A DES (choline chloride/citric acid) is reported for the first time to convert NAG to 3A5AF with a yield of 47.11 mol%.
Switchable product selectivity in dehydration of N-acetyl-D-glucosamine promoted by choline chloride-based deep eutectic solvents
[J].DOI:10.1016/j.isci.2023.106980 URL [本文引用: 1]
Direct conversion of chitin into a N-containing furan derivative
[J].DOI:10.1039/C3GC42436G URL [本文引用: 1]
Complete 1H and 13C NMR spectral assignment of D-glucofuranose
[J].DOI:10.1016/j.carres.2021.108477 URL [本文引用: 4]
Monosaccharide isomer interconversions become significant at high temperatures
[J].DOI:10.1021/acs.jpca.8b07217 URL [本文引用: 2]
Mechanism of the dehydration of D-fructose to 5-hydroxymethylfurfural in dimethyl sulfoxide at 150 ℃: an NMR study
[J].DOI:10.1016/j.carres.2008.09.008 URL [本文引用: 3]
3-Deoxy-glucosone is an intermediate in the formation of furfurals from D-glucose
[J].DOI:10.1002/cssc.v4.8 URL
Dehydration of D-fructose to hydroxymethylfurfural in sub- and supercritical fluids
[J].DOI:10.1016/j.supflu.2005.04.004 URL [本文引用: 1]
Sustainable solvent systems for use in tandem carbohydrate dehydration hydrogenation
[J].DOI:10.1021/sc400044d URL [本文引用: 1]
Molecular mapping of the acid catalysed dehydration of fructose
[J].DOI:10.1039/c2cc31689g URL [本文引用: 1]
β-directing effect of ionic liquid in mannopyranosylation: a potential access to stereoselective construction of the 1,2-cis-β-D-mannopyranosyl linkage
[J].
Gas-liquid chromatography of trimethylsilyl derivatives of sugars and related substances
[J].DOI:10.1021/ja00899a032 URL [本文引用: 1]
Ionic liquids and deep eutectic solvents: A brief prologue and their applications in sustainable extraction and separation processes
[J].DOI:10.1016/j.jics.2025.101638 URL [本文引用: 1]
Functionalized imidazolium ionic liquids-modified chitosan materials: From synthesis approaches to applications
[J].DOI:10.1016/j.reactfunctpolym.2023.105779 URL [本文引用: 1]
Basicity characterization of imidazolyl ionic liquids and their application for biomass dissolution
[J].
Dehydration of xylose to furfural over imidazolium-based ionic liquid with phase separation
[J].
DOI:10.3390/catal11121552
URL
[本文引用: 1]
An environmentally friendly catalyst and task-specific ionic liquid (IL), 1-(4-sulfonic acid) butyl-3-cetyl-2-methyl imidazolium hydrogen sulfate, was applied to the dehydration of xylose to furfural. Its structure was determined by FT-IR, 1H NMR technologies. The solubility of IL in water changed with the temperature, and had the advantages of homogeneous and heterogeneous catalysts. At the given conditions, xylose conversion of 95.3% and furfural yield of 67.5% were achieved over IL.
Design and synthesis of Brønsted-Lewis acidic tetraimidazolyl ionic liquids for efficient catalytic conversion of glucose to 5-hydroxymethylfurfural in water/1-octanol
[J].DOI:10.1016/j.apcata.2022.118981 URL [本文引用: 1]
Glucosamine condensation catalyzed by 1-ethyl-3-methylimidazolium acetate: mechanistic insight from NMR spectroscopy
[J].
DOI:10.1039/c5cp02169c
PMID:26278065
[本文引用: 4]
The basic ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2C1Im][OAc]) could efficiently catalyze the conversion of 2-amino-2-deoxy-d-glucose (GlcNH2) into deoxyfructosazine (DOF) and fructosazine (FZ). Mechanistic investigation by NMR studies disclosed that [C2C1Im][OAc], exhibiting strong hydrogen bonding basicity, could coordinate with the hydroxyl and amino groups of GlcNH2via the promotion of hydrogen bonding in bifunctional activation of substrates and further catalyzing product formation, based on which a plausible reaction pathway involved in this homogeneous base-catalyzed reaction was proposed. Hydrogen bonding as an activation force, therefore, is responsible for the remarkable selectivity and rate enhancement observed.
Conversion of chitin and N-acetyl-D-glucosamine into a N-containing furan derivative in ionic liquids
[J].DOI:10.1039/C5RA00382B URL [本文引用: 1]
Ternary deep eutectic solvents catalyzed D-glucosamine self-condensation to deoxyfructosazine: NMR study
[J].DOI:10.1016/j.gee.2020.04.010 URL [本文引用: 1]
Mechanism of the self-condensation of GlcNH2: insights from in situ NMR spectroscopy and DFT study
[J].DOI:10.1016/j.apcatb.2016.09.058 URL
Three-dimensional structures of 3/4/5-O-feruloylquinic acids by NMR spectroscopy and quantum chemistry calculation
[J].
核磁共振实验结合量子化学计算研究3/4/5-O-阿魏酰奎宁酸三维结构
[J].3/4/5-O-阿魏酰奎宁酸(3/4/5-O-feruloylquinic acid, 3/4/5-O-FQA)具有抗氧化活性,对预防多种疾病和维持人体健康有益,然而其准确三维结构尚不清楚.本文采用核磁共振波谱技术,结合量子化学理论计算方法,研究了3/4/5-O-FQA分别在气相、D<sub>2</sub>O溶液和DMSO-d<sub>6</sub>溶液中的三维结构.结果表明,3-O-FQA在D<sub>2</sub>O和DMSO-d<sub>6</sub>两种溶液中的三维结构存在差异,主要表现在酯化位H-3位置处二面角τ (H3C3O1C9的不同,相差4.147°.这些结构差异导致了3-O-FQA在两种溶液中的<sup>1</sup>H 核磁共振谱图中H-3质子裂分方式不同.该工作给出了3/4/5-O-FQA的精确结构信息,能够为进一步研究其性质和应用价值提供参考和帮助.
NMR data analysis of acarbose
[J].
阿卡波糖的核磁共振数据解析
[J].
DOI:10.11938/cjmr20243125
[本文引用: 1]
阿卡波糖是一种α-葡萄糖苷酶抑制剂,凭借其独特的作用机制,目前已被广泛应用于II型糖尿病的治疗中. 本文利用DEPT-135、<sup>1</sup>H-<sup>1</sup>H COSY、<sup>1</sup>H-<sup>13</sup>C HSQC和<sup>1</sup>H-<sup>13</sup>C HMBC等核磁共振(NMR)技术,以DMSO-d<sub>6</sub>为溶剂,对阿卡波糖的<sup>1</sup>H NMR、<sup>13</sup>C NMR信号进行了全归属,获得了在重水溶剂中无法获得的羟基<sup>1</sup>H NMR信号,补充了C环α、β双信号氢谱和碳谱数据,确证了其分子结构,为阿卡波糖药品安全及质量控制提供了可靠的数据基础.
Observation of the keto tautomer of D-fructose in D2O using 1H NMR spectroscopy
[J].DOI:10.1016/j.carres.2011.11.003 URL [本文引用: 1]
Isomeric distribution of monosaccharides in deep eutectic solvents: NMR study
[J].DOI:10.1016/j.molliq.2018.01.166 URL [本文引用: 2]
NMR studies of the tautomer distributions of D‑fructose in lower alcohols/DMSO‑d6
[J].DOI:10.1016/j.molliq.2018.09.067 URL [本文引用: 1]
Synthesis, characterization and catalysis performance of ionic liquid 1-butyl-3- methylimidazolium chlorozincate
[J].
1-丁基-3-甲基咪唑氯化锌离子液体的合成、表征及催化性能
[J].
Structural elucidation and quantitative analysis of hydrogenation products of anthracene by NMR spectroscopy
[J].
蒽加氢产物的结构指认和定量核磁共振分析
[J].
DOI:10.11938/cjmr20202849
[本文引用: 1]
本文利用多种液体核磁共振(NMR)技术,综合分析了在三个不同反应条件下蒽催化加氢反应获得的产物混合物.利用二维扩散排序谱(DOSY)和一维选择性激发谱(selTOCSY)确定了产物中含有的二氢蒽、四氢蒽、对称八氢蒽和非对称八氢蒽;利用<sup>1</sup>H NMR、<sup>13</sup>C NMR、DEPT135、<sup>1</sup>H-<sup>1</sup>H COSY、<sup>1</sup>H-<sup>13</sup>C HSQC实验对二氢蒽、四氢蒽和对称八氢蒽的<sup>1</sup>H和<sup>13</sup>C NMR信号进行了详细归属;利用定量核磁共振氢谱(QNMR)计算得到了蒽的转化率和产物的选择性.本研究可用于指导优化催化反应条件,提高产物对称八氢蒽的选择性,同时为稠环类芳烃催化加氢产物的分析提供系统的NMR技术方案.
Efficient production of 5-ethoxymethylfurfural from fructose by sulfonic mesostructured silica using DMSO as co-solvent
[J].DOI:10.1016/j.cattod.2016.02.016 URL [本文引用: 1]
Efficient catalytic system for the conversion of fructose into 5-ethoxymethylfurfural
[J].
DOI:10.1016/j.biortech.2013.02.110
PMID:23567707
[本文引用: 1]
DMSO can improve the selectivity of 5-hydroxymethylfurfural (HMF) in the conversion of carbohydrates. However, one of the bottlenecks in its application is product separation. Thus a one-pot synthesis of 5-ethoxymethylfurfural (EMF) rather than HMF from fructose in ethanol-DMSO was investigated. Phosphotungstic acid was used as an effective catalyst. The yield of EMF can be reached as high as 64% in the mixed solvent system of DMSO and ethanol within 130 min at 140 °C. Ethyl levulinate (LAE) was detected as the main by-product, the yield of which increased with the reaction time, temperature and the amount of catalyst. In addition, the existence of water could significantly reduce the yield of EMF and increased the yield of LAE. Most importantly, it was discovered that EMF could be much more efficiently extracted from the reaction solvent system by some organic solvents than HMF.Copyright © 2013 Elsevier Ltd. All rights reserved.
Pure shift NMR: application of 1D PSYCHE and 1D TOCSY-PSYCHE techniques for directly analyzing the mixtures from biomass-derived platform compound hydrogenation/hydrogenolysis
[J].DOI:10.1021/acssuschemeng.0c06882 URL [本文引用: 1]
Acyclic forms of [1-13C]aldohexoses in aqueous solution: quantitation by 13C NMR and deuterium isotope effects on tautomeric equilibria
[J].DOI:10.1021/jo010541m URL [本文引用: 2]
Pyranose-furanose and anomeric equilibria: influence of solvent and of partial methylation
[J].
DOI:10.1139/v66-304
URL
[本文引用: 4]
Sugars possessing the arabino (2,3,4-trans,cis) configuration exist as furanoses to a greater extent in dimethyl sulfoxide than in water. Their 2,3-di-O-methyl derivatives show an even stronger preference for a five membered ring structure in both solvents. This is most marked for 2,3-di-O-methyl-D-arabinose and 2,3-di-O-methyl-D-altrose, which are 65% and 80% furanose, respectively, in dimethyl sulfoxide. Compounds in the xylo (2,3,4-trans,trans) or lyxo (2,3,4-cis,trans) series show little tendency to be furanoses in either solvent. However, α-pyranoses in the lyxo series are relatively more stable in dimethyl sulfoxide than in water, whereas the anomeric composition for members of the xylo series is the same in both solvents. Some of these variations in the equilibria are attributed to the preferential stabilization of pyranose forms in water. D-Lyxose and D-ribose show nuclear magnetic resonance spectral differences in the two solvents that appear to be due to conformational, as well as tautomeric, equilibrium changes.An equatorial anomeric hydroxyl proton of a given pair in dimethyl sulfoxide shows a larger spacing (6.5–8.0 c.p.s.) than an axial anomeric hydroxyl proton (4–5 c.p.s.). The signal for the latter proton occurs at higher field than an equatorial OH-1, except when OH-2 is axial.
Structure and solution equilibria of D-glucose and D-mannose sulfite adducts
[J].An X-ray crystallographic study has confirmed that the potassium bisulfite adducts of D-glucose and D-mannose have open-chain structures with R and S configurations respectively at C-1. NMR studies have shown that each sugar gives rise to two bisulfite compounds, and solution-state structures and conformations of these isomers have been deduced from analysis of (1)H NMR spectra. (13)C NMR data for the four adducts are given. Furanose forms of the D-glucose and D-mannose have been detected in the equilibrium solutions.
Efficient 1H-NMR quantitation and investigation of N-acetyl-D-glucosamine (GlcNAc) and N,N'-diacetylchitobiose (GlcNAc)2 from chitin
[J].
DOI:10.3390/ijms12095828
URL
[本文引用: 1]
A quantitative determination method of N-acetyl-D-glucosamine (GlcNAc) and N,N'-diacetylchitobiose (GlcNAc)2 is proposed using a proton nuclear magnetic resonance experiment. N-acetyl groups of GlcNAc and (GlcNAc)2 are chosen as target signals, and the deconvolution technique is used to determine the concentration of the corresponding compound. Compared to the HPLC method, 1H-NMR spectroscopy is simple and fast. The method can be used for the analysis of chitin hydrolyzed products with real-time analysis, and for quantifying the content of products using internal standards without calibration curves. This method can be used to quickly evaluate chitinase activity. The temperature dependence of 1H-NMR spectra (VT-NMR) is studied to monitor the chemical shift variation of acetyl peak. The acetyl groups of products are involved in intramolecular H-bonding with the OH group on anomeric sites. The rotation of the acetyl group is closely related to the intramolecular hydrogen bonding pattern, as suggested by the theoretical data (molecular modeling).
Product distribution control for glucosamine condensation: nuclear magnetic resonance (NMR) investigation substantiated by density functional calculations
[J].DOI:10.1021/acs.iecr.6b05057 URL [本文引用: 1]
Studies on ketoses, 1 Distribution of furanoid and pyranoid tautomers of D-fructose in water, dimethyl sulfoxide, and pyridine via 1H NMR intensities of anomeric hydroxy groups in [d6]DMSO
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}