


引言
蛋白质作为生物大分子在生命活动中发挥着核心功能作用,其三级结构的精确解析及构象动态的深入研究对阐明生物学机制具有关键意义.经典的结构生物学研究方法,包括X射线晶体学(X-ray Crystallography)[1]、核磁共振(NMR)[2]以及冷冻电镜(Cryo-EM)[3],在静态结构解析领域已取得突破性进展.然而,这些技术体系存在显著的应用限制:X-ray依赖高质量晶体样本制备,难以表征溶液环境下的动态构象[4];NMR受限于分子量阈值及样品浓度要求[5];Cryo-EM虽在超大分子复合体结构解析中表现优异,但对小分子量及高动态性蛋白体系仍存在分辨率瓶颈[6].因此,发展能够在接近生理条件下研究蛋白质构象变化和动力学特性的技术显得尤为重要.
氢-氘交换(Hydrogen-Deuterium Exchange,HDX)是重要的蛋白质动态表征技术之一,近年来随着质谱(MS)和NMR仪器灵敏度的提高,其在结构生物学中的应用持续拓展.由于HDX速率与局部结构的溶剂可及性及氢键稳定性密切相关,动态区域或构象变化活跃的位点会更快发生交换,而稳定结构(如α螺旋、β折叠)中的酰胺氢因氢键保护交换速率较慢.通过定量分析主链酰胺氢与溶剂氘的交换速率,可解析蛋白质的构象柔韧性、配体结合位点及动态变构.早期氢氘交换研究主要依赖NMR检测(HDX-NMR)[7,8],随着MS灵敏度和数据解析算法的提升,HDX-MS也逐渐用于蛋白质的研究,尤其在抗体药物研发和膜蛋白研究中广泛应用[9,10].HDX实验通过检测酰胺氢的氘代程度及信号衰减,结合时间分辨谱图解析特定残基的动态信息,可定量分析主链酰胺氢与溶剂氘的交换速率,可间接揭示蛋白质的动态区域、结合界面及构象变化.总体来说,HDX-NMR技术可在近生理条件下对蛋白质进行无损检测,适用于溶液态天然构象的动态解析;而HDX-MS在较高通量和复杂体系分析中具有独特优势,二者在应用上各具特色.
1 HDX技术原理
图1
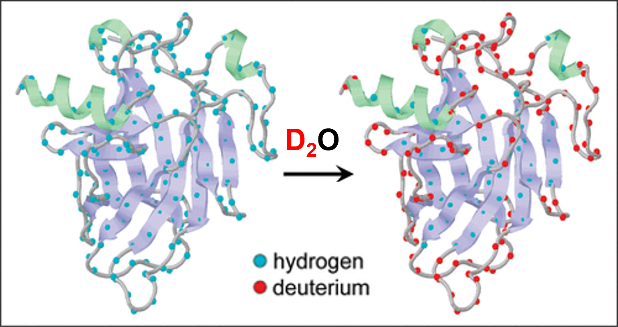
图1
HDX原理示意图[16]. 利用氢(H)与氘(D)两种同位素的化学交换行为,探测蛋白质分子中特定氢原子的可及性和动态特性
Fig. 1
Schematic illustration of the HDX principle[16]. The chemical exchange behavior between hydrogen (H) and deuterium (D) isotopes is utilized to probe the accessibility and dynamic properties of specific hydrogen atoms in protein molecules
2 HDX-NMR和HDX-MS的实验流程及技术发展
2.1 HDX-NMR实验流程
蛋白质分子的HDX-NMR实验,通常是对15N-标记的蛋白质样品采集2D ¹H-15N HSQC(Heteronuclear Single Quantum Coherence)谱,观测主链酰胺键上的1H-15N信号强度随H/D交换时间的衰减过程(图2). HDX-NMR实验包括以下步骤:常规NMR样品制备、HDX、HDX-NMR数据采集和数据分析.首先,制备15N-标记的蛋白质分子样品,优化NMR实验条件,包括缓冲溶液(pH、盐浓度、5%~10%的D2O用于锁场等)、样品浓度以及实验温度等,采集HDX前的2D 1H-15N HSQC谱;随后,将该样品进行低温真空冻干后,溶于与之前实验相同体积的D2O中(特殊情况下,比如醋酸铵缓冲溶液,还需要重新校正pH值),制备用于HDX-NMR的蛋白质样品.之后,采集不同HDX时间点的2D 1H-15N HSQC谱图(在交换过程早期,通常需要对HDX-NMR实验数据进行密集采集,比如每间隔10或15 min采一次样.交换时间达2~4 h后,可以增加采样的间隔时间,根据信号强度的变化程度采集所需交换时间的实验数据).对HDX-NMR实验数据进行分析,拟合峰强度随交换时间的衰减可以评估单个氨基酸酰胺氢的交换速率.常规的2D 1H-15N HSQC谱采样时间需要数分钟,因此HDX-NMR实验揭示了一段时间内的平均交换速率[8,15,19].采用快速采样的NMR脉冲序列,比如SOFAST-HMQC等(Bruker脉冲序列:sfhmqcf3gpph),可以将单次谱图采集时间减少到1 min以内,因而可以分析更短时间内的交换过程[20].此外,由于NMR方法固有的低灵敏度特性,超高场谱仪和超低温探头,将有助于提高HDX-NMR的灵敏度,缩短谱图采集时间.
图2
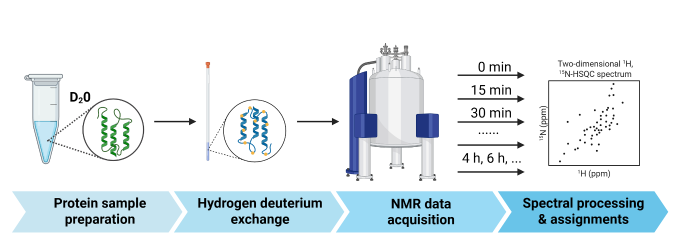
图2
HDX-NMR技术流程,主要包括以下步骤:蛋白样品制备、氢氘交换、NMR数据采集以及谱图处理与归属
Fig. 2
HDX-NMR workflow, including protein sample preparation, hydrogen-deuterium exchange, NMR data acquisition, spectral processing and assignments
2.2 HDX-NMR的优势与局限性
HDX-NMR融合了NMR技术的多种优势,在研究蛋白质动态结构与相互作用过程中,能够以残基分辨率提供结构变化信息,因而在解析高分辨率构象动态“交换”方面展现出独特且显著的优势[21,22].HDX-NMR将NMR的精准性与氢交换的时间窗口巧妙结合,可在近生理条件下以残基级分辨率逐位点解析蛋白质构象变化,为刻画高分辨率动态能量面提供了重要的手段.传统方法(化学变性、停流动力学、整体光谱)受限于微秒-毫秒时间尺度与高变性剂环境,只能报告体系平均行为,无法捕捉秒-小时级、隐藏在天然态盆地中的高能局部展开态.Basak等人利用HDX-NMR技术,在零变性剂条件下清晰显影了人工蛋白Di-III_14内部被传统技术忽略的粗糙能量面,阐明了其“极端耐尿素却又富集高能态”表观矛盾的根源,并揭示了静电-疏水网络如何协同调控βα折叠自由能面的复杂景观[23].
与HDX-MS相比,HDX-NMR的独特优势体现在:能在氨基酸水平解析蛋白质构象变化,提供更精确的位点信息;且NMR检测不破坏样品,适合长期跟踪蛋白结构变化;同时,其直接检测NMR活性核信号,无需蛋白酶消化,避免了样品制备中可能引入的人工偏差.然而,HDX-NMR的局限性也较为明显. 其一,时间分辨率限于秒级以上,难以捕捉毫秒级动态过程(如快速折叠中间体、酶催化过渡态),需与时间分辨质谱(如超快HDX-MS)等技术互补才能全面揭示动态机制.其二,NMR信号解析度随蛋白分子量增加而下降,通常难以研究分子量大于30 kDa的蛋白. 其三,实验对样品浓度要求较高(通常 > 100 µmol/L),对研究难高效表达或纯化的蛋白构成挑战.其四,部分氨基酸残基的氢交换速率过快或过慢,可能导致信号难以有效检测,影响数据完整性.其五,实验涉及氘交换、时间点控制、NMR数据采集与解析等多个复杂步骤,周期长且对技术操作要求高.综上,HDX-NMR在蛋白质动态结构研究中具有不可替代的优势,但其适用性受限于蛋白大小、样品浓度及实验复杂性.因此,实际应用中通常需结合HDX-MS或其他生物物理技术,以获取更全面的结构信息[7].
2.3 HDX-MS实验流程与数据处理
HDX-MS主要依托一系列精细化实验流程,实现对蛋白质结构及动力学特性的解析.首先,将蛋白样品溶解于使用重水(D2O)配制的缓冲液中,使可交换的酰胺氢逐步被氘取代.该氘标记过程的时间范围可依据实验需求设定,覆盖从秒级至小时级的不同时间尺度.随后,通过在低温(0~4 ℃)及低pH(~2.5)条件下迅速终止交换反应,以有效抑制氘-氢回交换,从而保持氘标记状态.接下来,采用胃蛋白酶进行在线酶解,确保肽段的高效生成,所得肽段经低温反相液相色谱(LC)分离,以降低回交换的影响.最终,利用高分辨率质谱(如Q-TOF、Orbitrap)监测各肽段的质荷比(m/z)变化,并据此推导蛋白质的HDX水平,从而解析其结构动态特征[11-
HDX-MS数据分析主要包括原始质谱数据的处理、氘交换程度的计算及蛋白质构象变化的解析(图3).首先,对原始质谱数据进行预处理,以去除噪声、校正质量漂移,并优化谱峰检测精度.随后,采用专业软件(如DynamX、HDExaminer或HX-Express)进行肽段识别与匹配,以确定不同时间点的氘交换水平. 氘交换程度的计算通常基于肽段的质荷比(m/z)变化,通过比较非氘化与氘化状态下的肽段质谱峰形,量化氘的占比.为提升空间分辨率,可结合高分辨率质谱技术或优化谱峰解析算法,以推测单个氨基酸水平的氘交换动力学[25,26].在数据可视化方面,常采用氘交换动力学曲线、热图(Heatmap)或差异交换图(difference plot)展示蛋白质构象变化.此外,为更直观地解析氘交换数据,HDX结果通常映射至蛋白质的三维结构,并结合分子动力学(MD)模拟或同源建模,以预测构象变化及其潜在机制.近年来,机器学习与人工智能(AI)技术的引入,为HDX-MS数据分析提供了新的策略.例如,深度学习算法可优化质谱峰检测,提高数据处理的准确性.基于人工智能的HDX(AI-HDX)方法能够依据蛋白质序列预测内在的构象动力学,其通过结合深度学习、实验HDX数据、序列比对及蛋白质结构预测,以揭示蛋白质的动态特性.AI-HDX在药物发现、蛋白质工程及生物医学研究等领域展现出广泛的应用前景[27].
图3

2.4 HDX-MS相关技术发展
HDX-MS在定位感兴趣区域时,其可靠性依赖于蛋白质序列的高覆盖度.然而,糖基化修饰是影响完整序列覆盖度的关键因素之一.在分析糖基化蛋白质时,糖肽往往因MS检测灵敏度较低或碎裂模式受限而丢失氘化信息,导致序列覆盖度下降.Haynes等利用电子转移低能碰撞诱导解离(EThcD)技术解析SARS-CoV-2刺突蛋白的糖肽,并通过计算糖肽的平均同位素质量偏移,确认糖肽已发生氘化.采用EThcD技术后,刺突蛋白的序列覆盖度由76%提高至84%,增强了结构重排区域的定位精度,提高了HDX-MS在复杂糖蛋白体系中的适用性和数据可靠性[30].
在蛋白质研究领域,HDX-MS技术借助前沿创新实现了重大突破.最新进展显示,通过采用先进的微流控芯片技术,并将温度降低至零下,HDX-MS成功克服了长期以来同位素回交换的难题.在低温环境下,蛋白质氘标记的丢失显著减少,使得该技术能够精准捕捉毫秒级的蛋白质动态变化,极大提升了时间分辨率,为研究短寿命构象态和快速的蛋白质相互作用提供了有力手段.在膜蛋白复合物解析方面,环形离子淌度技术的应用让HDX-MS取得了进一步发展.该技术将离子引入环形轨道,通过延长离子迁移路径,显著提升了分辨率.在分析膜蛋白复合物时,这一技术优势得以充分展现,与传统方法相比,肽段检出量提升了31%~222%,增幅显著.更多肽段的检出意味着能够获取更丰富的膜蛋白结构信息,有助于深入解析膜蛋白复合物的复杂结构与功能,推动膜蛋白相关研究迈向新高度[31,32].
2.5 HDX-MS与HDX-NMR的技术对比与互补性
HDX-MS与HDX-NMR是解析蛋白质动态构象的两种重要技术,其在技术特性及应用场景上存在显著差异(表1).HDX-MS以其较高的灵敏度优势,使其能够在低浓度蛋白样品条件下(低至纳摩尔级)获得可靠的数据,因此适用于多种蛋白质及其复合物的结构动力学研究[33].相比之下,HDX-NMR受限于较高的样品浓度需求(≥0.1 mmol/L)及较小的分子量范围(通常 < 30 kDa),更适用于高纯度的小型蛋白体系,以解析其精细的构象动态特征[8].在具体应用方面,HDX-MS在膜蛋白动力学[34]、抗体-抗原互作[35]及大分子复合物研究[36]中表现出独特优势,而HDX-NMR主要用于解析小型蛋白的变构机制、酶的折叠动力学及部分未折叠状态的构象特性[37].两种技术的互补性可通过联合应用进一步强化.
表1 HDX-NMR与HDX-MS对比
Table 1
| 比较维度 | HDX-NMR | HDX-MS |
|---|---|---|
| 技术原理 | HDX后蛋白质分子酰胺键上的H-N信号强度的衰减 | HDX后,酶解为肽段,通过MS检测质荷比变化 |
| 分辨率 | 单个氨基酸残基分辨率,定位精确 | 肽段级别分辨率,具体位点定位依赖酶解位点 |
| 结构信息 | 可提供构象变化、柔性区、相互作用位点等细节 | 可识别构象变化区域,但精细结构信息有限 |
| 实时动态监测 | 监测某一时间段的蛋白质分子信号的平均变化 | 需快速淬灭和冷冻,实时性相对较弱 |
| 样品处理 | 冻干重溶于重水,无需酶解,避免酶解引入偏差 | 需蛋白酶消化,可能引入序列覆盖度不全或选择性偏差 |
| 适用蛋白大小 | 分子量限制,通常适于 < 30 kDa的中小蛋白 | 可应用于大分子蛋白甚至多蛋白复合物 |
| 样品浓度要求 | 浓度要求高(通常 > 0.1 mmol/L) | 浓度要求低,适合高通量筛选 |
| 检测灵敏度 | 灵敏度相对较低 | MS灵敏度高,适合复杂样品和痕量检测 |
| 数据解析难度 | 谱图复杂,解析过程需要专业知识,周期较长 | 数据分析流程成熟,软件支持广泛,适合标准化处理 |
| 非破坏性 | 样品可重复使用,适合长期跟踪观察 | 酶解和MS过程不可逆,样品不可回收 |
| 典型应用 | 蛋白-配体结合、构象动态研究、弱相互作用分析 | 构象筛选、蛋白质稳定性、复合物交互界面快速分析 |
HDX-MS和HDX-NMR结合可用于蛋白质构象转变的精确研究,朊病毒蛋白(PrPᴄ)向错误折叠的Scrapie型(PrPˢᶜ)转换是传染性海绵状脑病的核心事件,构象柔性是其易转换的关键,研究者们利用HDX-MS和HDX-NMR,在pH = 4(错误折叠促进条件)下对全长小鼠朊病毒蛋白(moPrP (23-231))的天然态(N态)进行结构和能量表征,发现N态与至少两种部分展开形式(PUFs)处于平衡状态.这些PUFs通过N态的随机波动形成,具有不同的溶剂暴露表面积和稳定性,其中一种PUFs与错误折叠初始中间体结构相似.HDX-MS通过肽段水平分析实现高通量筛选,快速确定发生显著氘交换的区域(如β1、α1-β2环);HDX-NMR则在残基水平提供精确信息,鉴定34个慢交换酰胺氢的动力学特征(如ΔGop).两者结合既实现了全局扫描,又获得了原子分辨率的结构波动细节.该发现为理解朊病毒蛋白的淀粉样变倾向提供了关键依据,证实了部分展开中间体在朊病毒构象转换中的重要作用[38].
3 HDX-NMR技术在蛋白质科学中的应用
3.1 蛋白质折叠机制的层级解析
Uzawa团队将超快速HDX与二维核磁共振(2D NMR)结合,在毫秒级时间尺度揭示了脱辅基肌红蛋白(ApoMb)的折叠层级性[39].研究发现,ApoMb的折叠遵循特定顺序:A、G和H螺旋首先形成稳定的二级结构核心(5~10 ms),随后B螺旋部分参与(50~100 ms),最后E、F螺旋及环区逐步整合(秒级).早期中间体呈现局部稳定、螺旋含量接近天然态但三级结构松散的特征,支持“熔球态”模型.该工作揭示了蛋白质折叠由拓扑结构引导的分层级组装过程,为理解蛋白质从序列到三维结构的编码规则提供了原子水平的实验依据.
3.2 分子动力学与水可及性表征
针对不溶性生物大分子体系(如膜蛋白、多糖),Gelenter等开发了基于2H-13C固体核磁共振(ssNMR)的HDX分析方法[40].该方法能够精确表征纳秒至毫秒时间尺度的分子运动(如侧链旋转和结构域波动),并通过氘交换实验(H/D Exchange)有效识别溶剂暴露区域,揭示生物大分子的表面可及性及氢键网络特征.相较于传统溶液NMR,该技术特别适用于不溶性生物大分子体系(如膜蛋白和纤维素等)的研究,为解析复杂生物大分子的构象变化、水合作用机制及其功能相关性提供了重要的高分辨率研究手段,在结构生物学和生物材料领域具有广泛的应用价值.
3.3 动态构象的瞬时态捕捉技术
为突破传统HDX-NMR的时间分辨率限制,Kuwajima团队设计了一种基于二甲基亚砜(DMSO)淬灭的HDX-2D NMR联用策略[41].该方法通过DMSO快速淬灭HDX反应,有效捕获蛋白质动态构象变化的瞬时状态,结合二维NMR技术实现了蛋白质结构动态的高分辨率解析.该技术具有三个显著优势:(1)可精确测定毫秒级时间尺度的蛋白质动态构象变化;(2)保持蛋白质天然构象完整性;(3)适用于研究蛋白质折叠、构象转换及分子识别等动态过程.实验结果表明,该方法能有效揭示蛋白质功能相关的动态结构特征,为蛋白质动态结构与功能关系研究提供了新的技术手段.
3.4 包涵体错误折叠机制的高灵敏探测
针对传统技术难以解析的蛋白质聚集体系,Naser等人利用HDX-NMR的位点特异性优势,对超氧化物歧化酶(SOD)包涵体进行原子级结构表征[42].该技术具有三个显著特点:(1)高灵敏度,可检测包涵体中微弱的天然结构特征;(2)位点特异性,能精确识别不同结构域的动态差异;(3)时间分辨能力,可追踪从微秒到小时级的构象变化.研究发现,虽然SOD包涵体整体呈现结构异质性,但其核心区域仍保留与天然态相似的二级结构、氢键网络和功能位点构象,这一发现为理解蛋白质错误折叠机制和开发包涵体复性新策略提供了重要依据.
4 HDX-MS在生物医学研究中的应用
4.1 蛋白质动态构象解析
HDX-MS在蛋白质-核酸互作及蛋白动态构象解析研究中发挥了重要作用.例如,针对转录因子INSM1的研究,HDX-MS揭示了其在自由态下的高度动态构象特征(图4).由于ZF2与ZF3之间存在较长的连接区段,ZF1与ZF2结构较为柔性,表明INSM1可能通过结构重塑来适应不同的结合伙伴.此外,HDX-MS进一步验证了INSM1 ZF1-ZF5结构域与M2 dsDNA之间的直接相互作用,并发现两者结合主要集中在ZF2和ZF3区域[43].这一研究不仅阐明了INSM1在转录调控中的作用机制,还为锌指蛋白与DNA结合的动态特征提供了实验依据,进一步拓展了HDX-MS在蛋白-核酸复合物研究中的应用潜力.
图4

图4
HDX-MS揭示INSM1与核酸结合的高度动态构象特征[43]. (a) HDX-MS数据镜像图,对比了INSM1 ZF1-ZF5游离状态(Apo)与结合M2 DNA状态(Holo)所检测到的各肽段的相对氘摄取量;(b)将Apo状态的各肽段在1 min时的相对氘摄取量映射至INSM1 ZF1-ZF5结构上;(c) HDX-MS数据差异图,展示了Apo与Holo状态INSM1 ZF1-ZF5之间相对氘摄取量的变化;(d)氢氘交换60 min时,INSM1 ZF1-ZF5中Apo与Holo状态间存在显著差异的片段
Fig. 4
HDX-MS reveals the highly dynamic conformational characteristics of INSM1 in binding to nucleic acids[43]. (a) A mirror plot of HDX-MS data comparing the relative deuterium uptake for each peptide of free INSM1 ZF1-ZF5 (Apo) and INSM1 ZF1-ZF5 bound by M2 DNA (Holo); (b) The relative deuterium uptake for each peptide of Apo INSM1 ZF1-ZF5 at 1 min was mapped on the structure of INSM1 ZF1-ZF5; (c) A difference plot of HDX-MS data showing the changes in relative deuterium uptake between Apo and Holo INSM1 ZF1-ZF5; (d) The segments of INSM1 ZF1-ZF5 with significant changes between Apo and Holo states at 60 min
4.2 药物靶点筛选与抗体药物
HDX-MS在靶向药物开发领域具有重要的应用价值,尤其是在抗体药物和小分子抑制剂的筛选与优化过程中.免疫检查点抑制剂的开发是近年来抗肿瘤治疗的前沿方向,HDX-MS在PD-1/PD-L1抗体药物研究中起到了关键作用.例如,在帕博利珠单抗(Pembrolizumab)的优化过程中,HDX-MS被用于精确定位其互补决定区(CDR)与PD-1受体的结合表位,并指导抗体的人源化改造,以提高其亲和力与特异性[10].此外,在新冠病毒(SARS-CoV-2)中和抗体研究中,利用HDX-MS发现氧化应激能够降低受体结合域(RBD)从“down”状态到“up”状态的相对能量障碍,使这种构象变化更加有利,从而增强与宿主细胞受体ACE2的结合能力.这一发现揭示了SARS-CoV-2刺突糖蛋白在氧化应激下的结构动力学演变机制,为理解其在病毒进化中的作用提供了新的视角,并为开发针对刺突糖蛋白的抗病毒策略提供了理论依据[30].
4.3 蛋白折叠与错误折叠
在神经退行性疾病研究中,HDX-MS被广泛应用于解析蛋白错误折叠的动力学特性及其对小分子抑制剂的响应.例如,在阿尔茨海默病(AD)研究中,使用HDX-MS方法学结合正交生化方法,扩展了我们对纤维化过程中全长tau蛋白构象转变的理解,在纤维化过程中,N端和C端结构域仍然暴露于溶剂,从可溶性聚集体到纤维的氘化水平相似[44].在帕金森病(PD)研究中,将毫秒级HDX-MS数据与聚集动力学相关联,发现α-突触核蛋白的中心C末端残基是驱动其成核和聚集的关键区域.这一研究为理解内在无序蛋白的局部结构动力学与其功能特性之间的关系提供了新方法,并为当前关于单体α-突触核蛋白的局部化学环境与其构象系综偏倚之间关系的理解提供了新的细节[45].
5 HDX-MS结合其他技术的应用
5.1 HDX-MS与冷冻电镜(Cryo-EM)结合
HDX-MS与Cryo-EM结合可用于解析大分子复合体的动态构象变化.Cryo-EM以其高分辨率解析蛋白的静态结构,而HDX-MS提供了构象变化的动力学信息.例如,在钙调磷酸酶(CN)与III型磷脂酰肌醇4-激酶α(PI4KA)形成多蛋白复合体组装机制研究中,Shaw等人将cryo-EM、HDX-MS及Alphafold3建模技术相结合,揭示了CN通过PxIxIT和LxVP基序与复合体动态结合[46].在SARS-CoV-2感染机制研究中,Baggen等人利用X-ray、Cryo-EM和HDX-MS发现了TMEM106B的管腔结构域(LD)与SARS-CoV-2刺突的受体结合基序结合,并证明了TMEM106B促进刺突介导的合胞体形成,提示了TMEM106B在病毒融合中的作用[47].
5.2 HDX-MS与X-ray晶体学结合
X射线晶体学提供了蛋白质在结晶状态下的静态结构,而HDX-MS则可用于研究溶液态蛋白的动力学特征.Shah等人采用了X-ray和HDX-MS两种技术探究了表面活性剂5-环己基-1-戊基-β-D-麦芽糖苷(CYMAL-5)结合对细胞色素P450 2B4(CYP2B4)结构的影响[48].在CYP2B4的晶体结构中,CYMAL-5结合在靠近活性位点的外周口袋中,进一步通过HDX-MS发现表面活性剂的结合不仅改变了蛋白质的构象,还影响了其动力学特性,这种结构和动力学的变化可能与CYP2B4的功能调节有关,例如影响其底物结合和催化活性.Léger等人在研究钙调蛋白(CaM)与拮抗剂calmidazolium(CDZ)相互作用时的动力学和结构变化时,结合尺寸排阻色谱-小角X射线散射(SEC-SAXS)、X-ray、HDX-MS和NMR技术,对CaM与CDZ的复合物进行了分子表征,发现CDZ的结合诱导CaM的两个结构域发生从开放到闭合的构象重定向,并导致其结构元件的强烈稳定化[49].
5.3 HDX-MS与表面等离子共振(SPR)结合
SPR技术广泛用于测定蛋白-配体相互作用的亲和力(KD值),但无法提供结合界面的动态信息.HDX-MS可用于补充SPR数据,揭示蛋白-配体结合引起的变构效应.例如,Grauslund等人在研究两种抗NadA单克隆抗体(mAb 1C6和mAb 7F11)与NadA抗原的相互作用时,通过HDX-MS和表面等离子共振(SPR)技术来分析它们的表位和抗体互补决定区(paratope)的映射,以解释这两种抗体在杀菌活性上的差异[50]. HDX-MS显示两种mAb在与NadA结合后,其重链和轻链的多个互补决定区(CDRs)都发生了构象变化,表明它们通过不同的CDRs与NadA相互作用.SPR实验进一步发现mAb 1C6可以以更高的结合位点数(平均3个分子)与NadA三聚体结合,而mAb 7F11的结合位点数较低(平均1.5个分子),这种结合位点数的差异可能是导致两种mAb在杀菌活性上差异的原因之一.
6 总结与展望
HDX-NMR与HDX-MS在未来的发展方向各具特色,并将进一步互补,推动蛋白质结构动力学研究的深入.HDX-NMR由于其在高分辨率解析蛋白构象及氢键网络方面的独特优势——尤其在原子级精度追踪氢键断裂/形成过程、直接量化秒级构象波动、解析别构调控中的残基特异性动态等方面具有不可替代性,未来有望通过动态核极化(DNP)[51]、超低温探头技术等增强信号灵敏度[52],使其适用于低丰度蛋白或较大分子体系.此外,基于超高场核磁共振仪(> 1.2 GHz)的技术进步将提升化学位移分辨率,使得更精细的氘交换模式解析成为可能.尤其在研究蛋白质折叠核(Folding Core)形成、变构信号传递路径及短寿命中间态方面,HDX-NMR可提供远超MS的时空分辨率.另一方面,智能脉冲序列优化[53]及快速采样技术(非均匀采样(NUS)[54]、带选择激发-短时采样(BEST)[55]、共享瞬态快速采样-异核单量子相关谱(FAST-HSQC)[56]等)将加速数据采集并降低信号重叠问题,进一步降低HDX-NMR的“死”时间,使HDX-NMR在大分子复合物、膜蛋白、内在无序蛋白(IDPs)等复杂研究体系中展现更大潜力.特别是对于难以酶解或含有复杂翻译后修饰的功能区域(如激酶催化口袋、G蛋白偶联受体跨膜区),HDX-NMR可直接在天然状态下获得无干扰的动态信息.此外,HDX-NMR结合计算模拟(如分子动力学模拟和量子力学/分子力学混合计算),将有助于推测氘交换速率的微观机制,解析局部构象变化及其对功能的影响,建立从原子运动到生物功能的直接因果关联.
另一方面,HDX-MS因其高通量、低样品需求、宽适用范围的特点,在未来有望借助超高分辨率MS(如FT-ICR-MS和Orbitrap-MS)进一步提高数据解析能力,使得更精准的肽段级甚至单残基级氘交换信息成为可能,但受限于酶解步骤和离子化效率,其在实时动态及氢键网络直接解析方面仍依赖HDX-NMR的互补验证.同时,原位HDX-MS的发展将使其能够在更接近生理环境的条件下研究蛋白质的动态变化,而时间分辨超快HDX-MS(millisecond级)的发展将拓展其对短寿命构象态和瞬态相互作用的解析能力,与HDX-NMR形成多时间尺度覆盖的协同观测体系.此外,HDX-MS结合人工智能(AI)和深度学习,可自动解析MS数据、预测氘交换模式,并通过整合多模态数据(如冷冻电镜和小角散射技术)重建更准确的蛋白构象变化过程.
未来这两种技术将在多个场景深度结合:例如利用HDX-MS快速扫描药物结合引起的全局构象扰动,再通过HDX-NMR精确定位关键残基的动态重排;或在膜蛋白研究中,通过HDX-MS分析可溶结构域,配合HDX-NMR解析脂质包裹区域的构象响应.单分子HDX-MS(如单分子离子淌度HDX-MS)有望突破群体平均效应的限制,实现对单个蛋白质分子的氘交换轨迹追踪,而HDX-NMR通过同位素标记特定残基,可验证单分子动态的代表性,共同揭示蛋白质折叠路径的异质性,为药物靶标筛选等领域提供前所未有的解析能力.最终,两种技术的融合将建立从原子运动(NMR)到系统构象景观(MS)的完整动态图谱,成为精准设计变构药物、解析病理突变机制的核心工具.
综上所述,HDX-MS在蛋白质动态全景测绘中发挥基石作用,而HDX-NMR凭借其原子级精度解析氢键网络演化的能力(如变构信号传递中的残基特异性重排),成为破解超快动态密码的核心工具.二者的深度协同构建了动态研究新范式:HDX-MS高效锁定功能相关构象扰动区域后,HDX-NMR精准阐释关键位点(如变构通路节点、翻译后修饰热点)的动态机理,形成从全局构象扫描到原子运动解译的完整研究链条.未来,两种技术将在变构药物理性设计(MS筛选先导化合物→ NMR优化结合动力学)及构象疾病机制解析(MS追踪病理聚集态→ NMR揭示早期错误折叠)中开辟新路径,推动结构生物学进入“从构象快照到动态影像”的新纪元.
利益冲突
无
参考文献
Turning protein crystallisation from an art into a science
[J].Protein crystallisation has gained a new strategic and commercial relevance in the post-genomic era because of its pivotal role in structural genomics. Producing high-quality crystals has always been a bottleneck to structure determination and, with the advent of proteomics, this problem is becoming increasingly acute. The task of producing suitable crystals may be tackled using two approaches. The first relies on empirical techniques that are based mainly on trial and error, and what is perceived to be the 'art' of crystallisation. The second approach is aimed at gaining an understanding of the fundamental principles that govern crystallisation; this knowledge may be applied to design experimental methodology for producing high-quality crystals of medical and industrial interest.Copyright 2004 Elsevier Ltd.
NMR studies of structure and function of biological macromolecules (Nobel lecture)
[J].DOI:10.1002/anie.v42:29 URL [本文引用: 1]
Single-particle cryo-EM-How did it get here and where will it go
[J].
DOI:10.1126/science.aat4346
PMID:30166484
[本文引用: 1]

Cryo-electron microscopy, or simply cryo-EM, refers mainly to three very different yet closely related techniques: electron crystallography, single-particle cryo-EM, and electron cryotomography. In the past few years, single-particle cryo-EM in particular has triggered a revolution in structural biology and has become a newly dominant discipline. This Review examines the fascinating story of its start and evolution over the past 40-plus years, delves into how and why the recent technological advances have been so groundbreaking, and briefly considers where the technique may be headed in the future.Copyright © 2018, American Association for the Advancement of Science.
Dynamic personalities of proteins
[J].DOI:10.1038/nature06522 [本文引用: 1]
Quantitative dynamics and binding studies of the 20S proteasome by NMR
[J].DOI:10.1038/nature05512 [本文引用: 1]
Cryo-EM: A unique tool for the visualization of macromolecular complexity
[J].
DOI:10.1016/j.molcel.2015.02.019
PMID:26000851
[本文引用: 1]
3D cryo-electron microscopy (cryo-EM) is an expanding structural biology technique that has recently undergone a quantum leap progression in its achievable resolution and its applicability to the study of challenging biological systems. Because crystallization is not required, only small amounts of sample are needed, and because images can be classified in a computer, the technique has the potential to deal with compositional and conformational mixtures. Therefore, cryo-EM can be used to investigate complete and fully functional macromolecular complexes in different functional states, providing a richness of biological insight. In this review, we underlie some of the principles behind the cryo-EM methodology of single particle analysis and discuss some recent results of its application to challenging systems of paramount biological importance. We place special emphasis on new methodological developments that are leading to an explosion of new studies, many of which are reaching resolutions that could only be dreamed of just a couple of years ago. Copyright © 2015 Elsevier Inc. All rights reserved.
Protein structure prediction using residue-resolved protection factors from hydrogen-deuterium exchange NMR
[J].DOI:10.1016/j.str.2021.10.006 URL [本文引用: 2]
Hydrogen/deuterium exchange and nuclear magnetic resonance spectroscopy reveal dynamic allostery on multiple time scales in the serine protease thrombin
[J].
DOI:10.1021/acs.biochem.1c00277
PMID:34159782
[本文引用: 3]
A deeper understanding of how hydrogen/deuterium exchange mass spectrometry (HDX-MS) reveals allostery is important because HDX-MS can reveal allostery in systems that are not amenable to nuclear magnetic resonance (NMR) spectroscopy. We were able to study thrombin and its complex with thrombomodulin, an allosteric regulator, by both HDX-MS and NMR. In this Perspective, we compare and contrast the results from both experiments and from molecular dynamics simulations. NMR detects changes in the chemical environment around the protein backbone N-H bond vectors, providing residue-level information about the conformational exchange between distinct states. HDX-MS detects changes in amide proton solvent accessibility and H-bonding. Taking advantage of NMR relaxation dispersion measurements of the time scale of motions, we draw conclusions about the motions reflected in HDX-MS experiments. Both experiments detect allostery, but they reveal different components of the allosteric transition. The insights gained from integrating NMR and HDX-MS into thrombin dynamics enable a clearer interpretation of the evidence for allostery revealed by HDX-MS in larger protein complexes and assemblies that are not amenable to NMR.
Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry
[J].
DOI:10.1021/ac100962c
PMID:20518534
[本文引用: 1]
The study of membrane protein structure and enzymology has traditionally been hampered by the inherent insolubility of membrane proteins in aqueous environments and experimental challenges in emulating an in vivo lipid environment. Phospholipid bilayer nanodiscs have recently been shown to be of great use for the study of membrane proteins since they offer a controllable, stable, and monodisperse model membrane with a nativelike lipid bilayer. Here we report the integration of nanodiscs with hydrogen exchange (HX) mass spectrometry (MS) experiments, thereby allowing for analysis of the native conformation of membrane proteins. gamma-Glutamyl carboxylase (GGCX), an approximately 94 kDa transmembrane protein, was inserted into nanodiscs and labeled with deuterium oxide under native conditions. Analytical parameters including sample-handling and chromatographic separation were optimized to measure the incorporation of deuterium into GGCX. Coupling nanodisc technology with HX MS offers an effective approach for investigating the conformation and dynamics of membrane proteins in their native environment and is therefore capable of providing much needed insight into the function of membrane proteins.
Epitope and paratope mapping of PD-1/nivolumab by mass spectrometry-based hydrogen-deuterium exchange, cross-linking, and molecular docking
[J].
DOI:10.1021/acs.analchem.0c01291
PMID:32441507
[本文引用: 2]
Programmed cell death-1 (PD-1), an antigen co-receptor on cell surfaces, is one of the conspicuous immune checkpoints. Nivolumab, a monoclonal antibody therapeutic approved by the FDA, binds to PD-1 and efficiently blocks its pathways. In this study, an integrated approach was developed to map the epitope/paratope of PD-1/nivolumab. The approach includes hydrogen-deuterium exchange mass spectrometry (HDX-MS) followed by electron-transfer dissociation (ETD), chemical cross-linking, and molecular docking. HDX-ETD offers some binding-site characterization with amino acid resolution. Chemical cross-linking provides complementary information on one additional epitope (i.e., the BC-loop) and a potential paratope at the N-terminus of the heavy chain. Furthermore, cross-linking identifies another loop region (i.e., the C'D-loop) that undergoes a remote conformational change. The distance restraints derived from the cross-links enable building high-confidence models of PD-1/nivolumab, evaluated with respect to a resolved crystal structure. This integrated strategy is an opportunity to characterize comprehensively other antigen-antibody interactions, to enable the understanding of binding mechanisms, and to design future antibody therapeutics.
Hydrogen/deuterium exchange mass spectrometry and its application in the study of protein-protein interaction
[J].
氢氘交换质谱技术及其在蛋白质相互作用研究中的应用
[J].
Application progress of hydrogen deuterium exchange mass spectrometry in structural study of proteins and protein complexes
[J].
氢氘交换质谱技术在蛋白质和蛋白复合物结构研究中的应用进展
[J].
Application of hydrogen-deuterium exchange mass spectrometry technology in the field of protein drug research
[J].
氢氘交换质谱技术在蛋白质类药物研究领域的应用
[J].
Hydrogen exchange mass spectrometry for studying protein structure and dynamics
[J].
DOI:10.1039/c0cs00113a
PMID:21173980
[本文引用: 2]
Hydrogen/deuterium exchange (HDX) mass spectrometry (MS) has become a key technique for monitoring structural and dynamic aspects of proteins in solution. This approach relies on the fact that exposure of a protein to D(2)O induces rapid amide H → D exchange in disordered regions that lack stable hydrogen-bonding. Tightly folded elements are much more protected from HDX, resulting in slow isotope exchange that is mediated by the structural dynamics ("breathing motions") of the protein. MS-based peptide mapping is a well established technique for measuring the mass shifts of individual protein segments. This tutorial review briefly discusses basic fundamentals of HDX/MS, before highlighting a number of recent developments and applications. Gas phase fragmentation strategies represent a promising alternative to the traditional proteolysis-based approach, but experimentalists have to be aware of scrambling phenomena that can be encountered under certain conditions. Electron-based dissociation methods provide a solution to this problem. We also discuss recent advances that facilitate the applicability of HDX/MS to membrane proteins, and to the characterization of short-lived protein folding intermediates. It is hoped that this review will provide a starting point for novices, as well as a useful reference for practitioners, who require an overview of some recent trends in HDX/MS.
Using NMR-detected hydrogen-deuterium exchange to quantify protein stability in cosolutes, under crowded conditions in vitro and in cells
[J].
Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS
[J].
DOI:10.1021/ac901154s
PMID:19788312
[本文引用: 3]
Understanding as much as possible about proteins in the shortest amount of time has long been a goal of hydrogen exchange (HX) MS. Recent technological advances have led to improvements in the technique, but has this goal yet been achieved? (To listen to a podcast about this Feature, please go to the Analytical Chemistry Web site at pubs.acs.org/journal/ancham.).
Parallel post-translational modification scanning enhancing hydrogen-deuterium exchange-mass spectrometry coverage of key structural regions
[J].
DOI:10.1021/acs.analchem.9b01410
PMID:31082219
[本文引用: 1]
Hydrogen-deuterium exchange-mass spectrometry (HDXMS) is a powerful technology to characterize conformations and conformational dynamics of proteins and protein complexes. HDXMS has been widely used in the field of therapeutics for the development of protein drugs. Although sufficient sequence coverage is critical to the success of HDXMS, it is sometimes difficult to achieve. In this study, we developed a HDXMS data analysis strategy that includes parallel post-translational modification (PTM) scanning in HDXMS analysis. Using a membrane-delimited G protein-coupled receptor (vasopressin type 2 receptor; VR) and a cytosolic protein (Na/H exchanger regulatory factor-1; NHERF1) as examples, we demonstrate that this strategy substantially improves protein sequence coverage, especially in key structural regions likely including PTMs themselves that play important roles in protein conformational dynamics and function.
Research advances in hydrogen-deuterium exchange mass spectrometry for protein epitope mapping
[J].
DOI:10.1007/s00216-020-03091-9
PMID:33404742
[本文引用: 1]
With the development of biomedical technology, epitope mapping of proteins has become critical for developing and evaluating new protein drugs. The application of hydrogen-deuterium exchange for protein epitope mapping holds great potential. Although several reviews addressed the hydrogen-deuterium exchange, to date, only a few systematic reviews have focused on epitope mapping using this technology. Here, we introduce the basic principles, development history, and review research progress in hydrogen-deuterium exchange epitope mapping technology and discuss its advantages. We summarize the main hurdles in applying hydrogen-deuterium exchange epitope mapping technology, combined with relevant examples to provide specific solutions. We describe the epitope mapping of virus assemblies, disease-associated proteins, and polyclonal antibodies as examples of pattern introduction. Finally, we discuss the outlook of hydrogen-deuterium exchange epitope mapping technology. This review will help researchers studying protein epitopes to gain a more comprehensive understanding of this technology.
Specific protein-urea interactions
[J].
Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds
[J].We demonstrate for different protein samples that 2D 1H-15N correlation NMR spectra can be recorded in a few seconds of acquisition time using a new band-selective optimized flip-angle short-transient heteronuclear multiple quantum coherence experiment. This has enabled us to measure fast hydrogen-deuterium exchange rate constants along the backbone of a small globular protein fragment by real-time 2D NMR.
The Structural and dynamic insights into the Ala97Ser amyloidogenic mutation in transthyretin
[J].DOI:10.1002/asia.v20.4 URL [本文引用: 3]
The B domain of protein A retains residual structures in 6 M guanidinium chloride as revealed by hydrogen/deuterium-exchange NMR spectroscopy
[J].
DOI:10.1002/pro.4569
PMID:36659853
[本文引用: 1]
The characterization of residual structures persistent in unfolded proteins is an important issue in studies of protein folding, because the residual structures present, if any, may form a folding initiation site and guide the subsequent folding reactions. Here, we studied the residual structures of the isolated B domain (BDPA) of staphylococcal protein A in 6 M guanidinium chloride. BDPA is a small three-helix-bundle protein, and until recently its folding/unfolding reaction has been treated as a simple two-state process between the native and the fully unfolded states. We employed a dimethylsulfoxide (DMSO)-quenched hydrogen/deuterium (H/D)-exchange 2D NMR techniques with the use of spin desalting columns, which allowed us to investigate the H/D-exchange behavior of individually identified peptide amide (NH) protons. We obtained H/D-exchange protection factors of the 21 NH protons that form an α-helical hydrogen bond in the native structure, and the majority of these NH protons were significantly protected with a protection factor of 2.0-5.2 in 6 M guanidinium chloride, strongly suggesting that these weakly protected NH protons form much stronger hydrogen bonds under native folding conditions. The results can be used to deduce the structure of an early folding intermediate, when such an intermediate is shown by other methods. Among three native helical regions, the third helix in the C-terminal side was highly protected and stabilized by side-chain salt bridges, probably acting as the folding initiation site of BDPA. The present results are discussed in relation to previous experimental and computational findings on the folding mechanisms of BDPA. This article is protected by copyright. All rights reserved.© 2023 The Protein Society.
Networks of electrostatic and hydrophobic interactions modulate the complex folding free energy surface of a designed βα protein
[J].
DOI:10.1073/pnas.1818744116
URL
[本文引用: 1]
Natural protein sequences have evolved to fold efficiently to their functional forms, traversing a relatively smooth energy surface with minimal frustration. The optimization of stability in de novo-designed proteins, without regard to dynamical processes required for function, can have unintended consequences on energy surfaces. Electrostatic and hydrophobic networks of side chains for Di-III_14, a small βα protein with a natural topology, create a rough energy surface whose high-energy states reflect the progressive unlocking of a tightly packed interior. Unbiased explorations of sequence space can reveal complex properties not seen in natural proteins, expanding our understanding of the relationships among sequence, structure, folding, and function of these ubiquitous biopolymers.
Hydrogen-deuterium exchange mass spectrometry for probing changes in conformation and dynamics of proteins
[J].
Challenges in the interpretation of protein h/d exchange data: a molecular dynamics simulation perspective
[J].
DOI:10.1021/acs.biochem.5b00215
PMID:25860179
[本文引用: 1]
Many protein structural investigations involve the use of H/D exchange (HDX) techniques. It is commonly thought that amide backbone protection arises from intramolecular H-bonding and/or burial of NH sites. Recently, fundamental HDX-related tenets have been called into question. The current work focuses on ubiquitin for exploring the defining features that distinguish amides in "open" (exchange-competent) and "closed" (exchange-incompetent) environments. Instead of relying on static X-ray structures, we employ all-atom molecular dynamics (MD) simulations for obtaining a dynamic view of the protein ground state and its surrounding solvent. The HDX properties for 57 out of 72 NH sites can be readily explained on the basis of backbone and side chain H-bonding, as well as solvent accessibility considerations. Unexpectedly, the same criteria fail for predicting the HDX characteristics of the remaining 15 amides. Significant protection is seen for numerous exposed NH sites that are not engaged in intramolecular H-bonds, whereas other amides that seemingly share the same features are unprotected. We scrutinize the proposal that H-bonding to crystallographically defined water can cause the protection of surface amides. For ubiquitin, the positioning of crystal water is not compatible with this idea. To further explore possible solvation effects, we tested for the presence of partially immobilized water networks. Our MD data reveal no difference in the solvation properties of protected vs unprotected surface amides, making it unlikely that restricted water dynamics can cause anomalous amide protection. The findings reported here suggest that efforts to deduce protein structural features on the basis of HDX protection factors may yield misleading results. This conclusion is relevant for initiatives that rely on sparse structural data as constraints for elucidating protein conformations. It may be necessary to pursue detailed quantum mechanical studies of the protein, the solvent, and the hydroxide catalyst for obtaining a comprehensive understanding of the factors that govern HDX rates. The considerable size of the systems involved makes such endeavors a daunting task.
Improved protein hydrogen/deuterium exchange mass spectrometry platform with fully automated data processing
[J].
DOI:10.1021/ac300535r
PMID:22571272
[本文引用: 1]
Protein hydrogen/deuterium exchange (HDX) followed by protease digestion and mass spectrometric (MS) analysis is accepted as a standard method for studying protein conformation and conformational dynamics. In this article, an improved HDX MS platform with fully automated data processing is described. The platform significantly reduces systematic and random errors in the measurement by introducing two types of corrections in HDX data analysis. First, a mixture of short peptides with fast HDX rates is introduced as internal standards to adjust the variations in the extent of back exchange from run to run. Second, a designed unique peptide (PPPI) with slow intrinsic HDX rate is employed as another internal standard to reflect the possible differences in protein intrinsic HDX rates when protein conformations at different solution conditions are compared. HDX data processing is achieved with a comprehensive HDX model to simulate the deuterium labeling and back exchange process. The HDX model is implemented into the in-house developed software MassAnalyzer and enables fully unattended analysis of the entire protein HDX MS data set starting from ion detection and peptide identification to final processed HDX output, typically within 1 day. The final output of the automated data processing is a set (or the average) of the most possible protection factors for each backbone amide hydrogen. The utility of the HDX MS platform is demonstrated by exploring the conformational transition of a monoclonal antibody by increasing concentrations of guanidine.
Artificial intelligence-based HDX (AI-HDX) prediction reveals fundamental characteristics to protein dynamics: Mechanisms on SARS-CoV-2 immune escape
[J].DOI:10.1016/j.isci.2023.106282 URL [本文引用: 1]
UV photodissociation mass spectrometry accurately localize sites of backbone deuteration in peptides
[J].
DOI:10.1021/acs.analchem.7b04683
PMID:29266933
[本文引用: 1]
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is now a routinely used technique to inform on protein structure, dynamics, and interactions. Localizing the incorporated deuterium content on a single residue basis increases the spatial resolution of this technique enabling detailed structural analysis. Here, we investigate the use of ultraviolet photodissociation (UVPD) at 213 nm to measure deuterium levels at single residue resolution in HDX-MS experiments. Using a selectively labeled peptide, we show that UVPD occurs without H/D scrambling as the peptide probe accurately retains its solution-phase deuterium labeling pattern. Our results indicate that UVPD provides an attractive alternative to electron mediated dissociation for increasing the spatial resolution of the HDX-MS experiment, capable of yielding high fragmentation efficiency, high fragment ion diversity, and low precursor ion charge-state dependency.
Top-down hydrogen-deuterium exchange analysis of protein structures using ultraviolet photodissociation
[J].
DOI:10.1021/acs.analchem.7b03655
PMID:29336549
[本文引用: 1]
Top-down hydrogen-deuterium exchange (HDX) analysis using electron capture or transfer dissociation Fourier transform mass spectrometry (FTMS) is a powerful method for the analysis of secondary structure of proteins in solution. The resolution of the method is a function of the degree of fragmentation of backbone bonds in the proteins. While fragmentation is usually extensive near the N- and C-termini, electron capture (ECD) or electron transfer dissociation (ETD) fragmentation methods sometimes lack good coverage of certain regions of the protein, most often in the middle of the sequence. Ultraviolet photodissociation (UVPD) is a recently developed fast-fragmentation technique, which provides extensive backbone fragmentation that can be complementary in sequence coverage to the aforementioned electron-based fragmentation techniques. Here, we explore the application of electrospray ionization (ESI)-UVPD FTMS on an Orbitrap Fusion Lumos Tribrid mass spectrometer to top-down HDX analysis of proteins. We have incorporated UVPD-specific fragment-ion types and fragment-ion mixtures into our isotopic envelope fitting software (HDX Match) for the top-down HDX analysis. We have shown that UVPD data is complementary to ETD, thus improving the overall resolution when used as a combined approach.
Inclusion of deuterated glycopeptides provides increased sequence coverage in hydrogen/deuterium exchange mass spectrometry analysis of SARS-CoV-2 spike glycoprotein
[J].DOI:10.1002/rcm.v38.5 URL [本文引用: 2]
Development of a thiol-ene microfluidic chip for hydrogen/deuterium exchange mass spectrometry (HDX-MS)
[J].DOI:10.1021/acs.analchem.4c06230 URL [本文引用: 1]
Cyclic ion mobility for hydrogen/deuterium exchange-mass spectrometry applications
[J].
DOI:10.1021/acs.analchem.3c05753
PMID:38561318
[本文引用: 1]
Hydrogen/deuterium exchange-mass spectrometry (HDX-MS) has emerged as a powerful tool to probe protein dynamics. As a bottom-up technique, HDX-MS provides information at peptide-level resolution, allowing structural localization of dynamic changes. Consequently, the HDX-MS data quality is largely determined by the number of peptides that are identified and monitored after deuteration. Integration of ion mobility (IM) into HDX-MS workflows has been shown to increase the data quality by providing an orthogonal mode of peptide ion separation in the gas phase. This is of critical importance for challenging targets such as integral membrane proteins (IMPs), which often suffer from low sequence coverage or redundancy in HDX-MS analyses. The increasing complexity of samples being investigated by HDX-MS, such as membrane mimetic reconstituted and IMPs, has generated need for instrumentation with greater resolving power. Recently, Giles et al. developed cyclic ion mobility (cIM), an IM device with racetrack geometry that enables scalable, multipass IM separations. Using one-pass and multipass cIM routines, we use the recently commercialized SELECT SERIES Cyclic IM spectrometer for HDX-MS analyses of four detergent solubilized IMP samples and report its enhanced performance. Furthermore, we develop a novel processing strategy capable of better handling multipass cIM data. Interestingly, use of one-pass and multipass cIM routines produced unique peptide populations, with their combined peptide output being 31 to 222% higher than previous generation SYNAPT G2-Si instrumentation. Thus, we propose a novel HDX-MS workflow with integrated cIM that has the potential to enable the analysis of more complex systems with greater accuracy and speed.
Investigation of the solid-state interactions in lyophilized human G-CSF using hydrogen-deuterium exchange mass spectrometry
[J].DOI:10.1021/acs.molpharmaceut.3c01211 URL [本文引用: 1]
Structural predictions of the functions of membrane proteins from HDX-MS
[J].
DOI:10.1042/BST20190880
URL
[本文引用: 1]
HDX-MS has emerged as a powerful tool to interrogate the structure and dynamics of proteins and their complexes. Recent advances in the methodology and instrumentation have enabled the application of HDX-MS to membrane proteins. Such targets are challenging to investigate with conventional strategies. Developing new tools are therefore pertinent for improving our fundamental knowledge of how membrane proteins function in the cell. Importantly, investigating this central class of biomolecules within their native lipid environment remains a challenge but also a key goal ahead. In this short review, we outline recent progresses in dissecting the conformational mechanisms of membrane proteins using HDX-MS. We further describe how the use of computational strategies can aid the interpretation of experimental data and enable visualisation of otherwise intractable membrane protein states. This unique integration of experiments with computations holds significant potential for future applications.
Epitope mapping of polyclonal antibodies by hydrogen-deuterium exchange mass spectrometry (HDX-MS)
[J].
DOI:10.1021/acs.analchem.1c00696
PMID:34308633
[本文引用: 1]
Epitope mapping of antibodies (Abs) is crucial for understanding adaptive immunity, as well as studying the mode of action of therapeutic antibodies and vaccines. Especially insights into the binding of the entire polyclonal antibody population (pAb) raised upon vaccination would be of unique value to vaccine development. However, very few methods for epitope mapping can tolerate the complexity of a pAb sample. Here we show how hydrogen-deuterium exchange mass spectrometry (HDX-MS) can be used to map epitopes recognized by pAb samples. Our approach involves measuring the HDX of the antigen in absence or presence of varied amounts of pAbs, as well as dissociating additives. We apply the HDX-MS workflow to pAbs isolated from rabbit immunized with factor H-binding protein (fHbp), a vaccine antigen. We identify four immunogenic regions located on the N- and C-terminal region of fHbp and provide insights into the relative abundance and avidity of epitope binding Abs present in the sample. Overall, our results show that HDX-MS can provide a unique and relatively fast method for revealing the binding impact of the entire set of pAbs present in blood samples after vaccination. Such information provides a rare view into effective immunity and can guide the design of improved vaccines against viruses or bacteria.
Structural dynamics in the evolution of SARS-CoV-2 spike glycoprotein
[J].
DOI:10.1038/s41467-023-36745-0
PMID:36918534
[本文引用: 1]
SARS-CoV-2 spike glycoprotein mediates receptor binding and subsequent membrane fusion. It exists in a range of conformations, including a closed state unable to bind the ACE2 receptor, and an open state that does so but displays more exposed antigenic surface. Spikes of variants of concern (VOCs) acquired amino acid changes linked to increased virulence and immune evasion. Here, using HDX-MS, we identified changes in spike dynamics that we associate with the transition from closed to open conformations, to ACE2 binding, and to specific mutations in VOCs. We show that the RBD-associated subdomain plays a role in spike opening, whereas the NTD acts as a hotspot of conformational divergence of VOC spikes driving immune evasion. Alpha, beta and delta spikes assume predominantly open conformations and ACE2 binding increases the dynamics of their core helices, priming spikes for fusion. Conversely, substitutions in omicron spike lead to predominantly closed conformations, presumably enabling it to escape antibodies. At the same time, its core helices show characteristics of being pre-primed for fusion even in the absence of ACE2. These data inform on SARS-CoV-2 evolution and omicron variant emergence.© 2023. The Author(s).
The knotted protein UCH-L1 exhibits partially unfolded forms under native conditions that share common structural features with its kinetic folding intermediates
[J].DOI:10.1016/j.jmb.2016.04.002 URL [本文引用: 1]
Partially unfolded forms of the prion protein populated under misfolding-promoting conditions: characterization by hydrogen exchange mass spectrometry and NMR
[J].
DOI:10.1074/jbc.M115.677575
PMID:26306043
[本文引用: 1]
The susceptibility of the cellular prion protein (PrP(C)) to convert to an alternative misfolded conformation (PrP(Sc)), which is the key event in the pathogenesis of prion diseases, is indicative of a conformationally flexible native (N) state. In the present study, hydrogen-deuterium exchange (HDX) in conjunction with mass spectrometry and nuclear magnetic resonance spectroscopy were used for the structural and energetic characterization of the N state of the full-length mouse prion protein, moPrP(23-231), under conditions that favor misfolding. The kinetics of HDX of 34 backbone amide hydrogens in the N state were determined at pH 4. In contrast to the results of previous HDX studies on the human and Syrian hamster prion proteins at a higher pH, various segments of moPrP were found to undergo different extents of subglobal unfolding events at pH 4, a pH at which the protein is known to be primed to misfold to a β-rich conformation. No residual structure around the disulfide bond was observed for the unfolded state at pH 4. The N state of the prion protein was observed to be at equilibrium with at least two partially unfolded forms (PUFs). These PUFs, which are accessed by stochastic fluctuations of the N state, have altered surface area exposure relative to the N state. One of these PUFs resembles a conformation previously implicated to be an initial intermediate in the conversion of monomeric protein into misfolded oligomer at pH 4. © 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR
[J].
DOI:10.1073/pnas.0804033105
URL
[本文引用: 1]
The earliest steps in the folding of proteins are complete on an extremely rapid time scale that is difficult to access experimentally. We have used rapid-mixing quench-flow methods to extend the time resolution of folding studies on apomyoglobin and elucidate the structural and dynamic features of members of the ensemble of intermediate states that are populated on a submillisecond time scale during this process. The picture that emerges is of a continuum of rapidly interconverting states. Even after only 0.4 ms of refolding time a compact state is formed that contains major parts of the A, G, and H helices, which are sufficiently well folded to protect amides from exchange. The B, C, and E helix regions fold more slowly and fluctuate rapidly between open and closed states as they search docking sites on this core; the secondary structure in these regions becomes stabilized as the refolding time is increased from 0.4 to 6 ms. No further stabilization occurs in the A, G, H core at 6 ms of folding time. These studies begin to time-resolve a progression of compact states between the fully unfolded and native folded states and confirm the presence an ensemble of intermediates that interconvert in a hierarchical sequence as the protein searches conformational space on its folding trajectory.
(2)H-(13)C correlation solid-state NMR for investigating dynamics and water accessibilities of proteins and carbohydrates
[J].DOI:10.1007/s10858-017-0124-7 [本文引用: 1]
DMSO-quenched H/D-exchange 2D NMR spectroscopy and its applications in protein science
[J].
DOI:10.3390/molecules27123748
URL
[本文引用: 1]
Hydrogen/deuterium (H/D) exchange combined with two-dimensional (2D) NMR spectroscopy has been widely used for studying the structure, stability, and dynamics of proteins. When we apply the H/D-exchange method to investigate non-native states of proteins such as equilibrium and kinetic folding intermediates, H/D-exchange quenching techniques are indispensable, because the exchange reaction is usually too fast to follow by 2D NMR. In this article, we will describe the dimethylsulfoxide (DMSO)-quenched H/D-exchange method and its applications in protein science. In this method, the H/D-exchange buffer is replaced by an aprotic DMSO solution, which quenches the exchange reaction. We have improved the DMSO-quenched method by using spin desalting columns, which are used for medium exchange from the H/D-exchange buffer to the DMSO solution. This improvement has allowed us to monitor the H/D exchange of proteins at a high concentration of salts or denaturants. We describe methodological details of the improved DMSO-quenched method and present a case study using the improved method on the H/D-exchange behavior of unfolded human ubiquitin in 6 M guanidinium chloride.
High-resolution NMR H/D exchange of human superoxide dismutase inclusion bodies reveals significant native features despite structural heterogeneity
[J].
Structural insights into a highly flexible zinc finger module unravel INSM1 function in transcription regulation
[J].DOI:10.1038/s41467-025-57478-2 [本文引用: 3]
Probing conformational dynamics of tau protein by hydrogen/deuterium exchange mass spectrometry
[J].DOI:10.1007/s13361-017-1815-8 URL [本文引用: 1]
Millisecond hydrogen/deuterium-exchange mass spectrometry approach to correlate local structure and aggregation in α-synuclein
[J].DOI:10.1021/acs.analchem.2c03183 URL [本文引用: 1]
Structure of calcineurin bound to PI4KA reveals dual interface in both PI4KA and FAM126A
[J].DOI:10.1016/j.str.2024.08.007 URL [本文引用: 1]
TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry
[J].
DOI:10.1016/j.cell.2023.06.005
PMID:37421949
[本文引用: 1]
SARS-CoV-2 is associated with broad tissue tropism, a characteristic often determined by the availability of entry receptors on host cells. Here, we show that TMEM106B, a lysosomal transmembrane protein, can serve as an alternative receptor for SARS-CoV-2 entry into angiotensin-converting enzyme 2 (ACE2)-negative cells. Spike substitution E484D increased TMEM106B binding, thereby enhancing TMEM106B-mediated entry. TMEM106B-specific monoclonal antibodies blocked SARS-CoV-2 infection, demonstrating a role of TMEM106B in viral entry. Using X-ray crystallography, cryogenic electron microscopy (cryo-EM), and hydrogen-deuterium exchange mass spectrometry (HDX-MS), we show that the luminal domain (LD) of TMEM106B engages the receptor-binding motif of SARS-CoV-2 spike. Finally, we show that TMEM106B promotes spike-mediated syncytium formation, suggesting a role of TMEM106B in viral fusion. Together, our findings identify an ACE2-independent SARS-CoV-2 infection mechanism that involves cooperative interactions with the receptors heparan sulfate and TMEM106B.Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Effect of detergent binding on cytochrome P450 2B4 structure as analyzed by X-ray crystallography and deuterium-exchange mass spectrometry
[J].
DOI:S0301-4622(16)30132-6
PMID:27280734
[本文引用: 1]
Multiple crystal structures of CYP2B4 have demonstrated the binding of the detergent 5-cyclohexyl-1-pentyl-β-D-maltoside (CYMAL-5) in a peripheral pocket located adjacent to the active site. To explore the consequences of detergent binding, X-ray crystal structures of the peripheral pocket mutant CYP2B4 F202W were solved in the presence of hexaethylene glycol monooctyl ether (C8E6) and CYMAL-5. The structure in the presence of CYMAL-5 illustrated a closed conformation indistinguishable from the previously solved wild-type. In contrast, the F202W structure in the presence of C8E6 revealed a detergent molecule that coordinated the heme-iron and extended to the protein surface through the substrate access channel 2f. Despite the overall structural similarity of these detergent complexes, remarkable differences were observed in the A, A', and H helices, the F-G cassette, the C-D and β4 loop region. Hydrogen-deuterium exchange mass spectrometry (DXMS) was employed to probe these differences and to test the effect of detergents in solution. The presence of either detergent increased the H/D exchange rate across the plastic regions, and the results obtained by DXMS in solution were consistent in general with the relevant structural snapshots. The study provides insight into effect of detergent binding and the interpretation of associated conformational dynamics of CYP2B4.Copyright © 2016 Elsevier B.V. All rights reserved.
Dynamics and structural changes of calmodulin upon interaction with the antagonist calmidazolium
[J].
DOI:10.1186/s12915-022-01381-5
PMID:35945584
[本文引用: 1]
Calmodulin (CaM) is an evolutionarily conserved eukaryotic multifunctional protein that functions as the major sensor of intracellular calcium signaling. Its calcium-modulated function regulates the activity of numerous effector proteins involved in a variety of physiological processes in diverse organs, from proliferation and apoptosis, to memory and immune responses. Due to the pleiotropic roles of CaM in normal and pathological cell functions, CaM antagonists are needed for fundamental studies as well as for potential therapeutic applications. Calmidazolium (CDZ) is a potent small molecule antagonist of CaM and one the most widely used inhibitors of CaM in cell biology. Yet, CDZ, as all other CaM antagonists described thus far, also affects additional cellular targets and its lack of selectivity hinders its application for dissecting calcium/CaM signaling. A better understanding of CaM:CDZ interaction is key to design analogs with improved selectivity. Here, we report a molecular characterization of CaM:CDZ complexes using an integrative structural biology approach combining SEC-SAXS, X-ray crystallography, HDX-MS, and NMR.We provide evidence that binding of a single molecule of CDZ induces an open-to-closed conformational reorientation of the two domains of CaM and results in a strong stabilization of its structural elements associated with a reduction of protein dynamics over a large time range. These CDZ-triggered CaM changes mimic those induced by CaM-binding peptides derived from physiological protein targets, despite their distinct chemical natures. CaM residues in close contact with CDZ and involved in the stabilization of the CaM:CDZ complex have been identified.Our results provide molecular insights into CDZ-induced dynamics and structural changes of CaM leading to its inhibition and open the way to the rational design of more selective CaM antagonists. Calmidazolium is a potent and widely used inhibitor of calmodulin, a major mediator of calcium-signaling in eukaryotic cells. Structural characterization of calmidazolium-binding to calmodulin reveals that it triggers open-to-closed conformational changes similar to those induced by calmodulin-binding peptides derived from enzyme targets. These results provide molecular insights into CDZ-induced dynamics and structural changes of CaM leading to its inhibition and open the way to the rational design of more selective CaM antagonists.© 2022. The Author(s).
Epitope and paratope mapping by HDX-MS combined with SPR elucidates the difference in bactericidal activity of two anti-NadA monoclonal antibodies
[J].DOI:10.1021/jasms.0c00431 URL [本文引用: 1]
Recent developments in materials and applications of triplet dynamic nuclear polarization
[J].DOI:10.1016/j.pnmrs.2024.05.001 URL [本文引用: 1]
400 MHz/263 GHz ultra-low temperature MAS-DNP using a closed-cycle helium gas cooling system and a solid-state microwave source
[J].DOI:10.1016/j.jmr.2025.107842 URL [本文引用: 1]
NMR spectral editing, water suppression, and dipolar decoupling in low-field NMR spectroscopy using optimal control pulses and multiple-pulse sequence
[J].DOI:10.1021/acs.analchem.3c05226 URL [本文引用: 1]
NUS-tool: A unified program for generating and analyzing sample schedules for nonuniformly sampled NMR experiments
[J].DOI:10.1016/j.jmr.2023.107458 URL [本文引用: 1]
An efficient combination of BEST and NUS methods in multidimensional NMR spectroscopy for high throughput analysis of proteins
[J].DOI:10.1039/C8RA00527C URL [本文引用: 1]
ASAP-HSQC-TOCSY for fast spin system identification and extraction of long-range couplings
[J].
DOI:S1090-7807(18)30351-3
PMID:30711785
[本文引用: 1]
Based on Ernst-angle-type excitation and Acceleration by Sharing Adjacent Polarization (ASAP), a fast HSQC-TOCSY experiment is introduced. In the approach, the DIPSI-2 isotropic mixing period of the ASAP-HSQC is simply shifted, which provides a TOCSY period without additional application of rf-energy. The ASAP-HSQC-TOCSY allows the acquisition of a conventional 2D in about 30 s. Alternatively, it allows the acquisition of highly carbon-resolved spectra (several Hz digital resolution) on the order of minutes. An ASAP-HSQC-TOCSY-IPAP variant, finally, allows the sign-sensitive extraction of heteronuclear long-range coupling constants from a pair of highly resolved spectra in less than an hour. Pulse sequences, several example spectra, and a discussion of results are given.Copyright © 2019 Elsevier Inc. All rights reserved.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}