


引言
蛋白质的氧化修饰包括可逆的修饰和不可逆的修饰两大类[1,2]. 可逆的修饰主要与细胞正常的生理过程有关,不会对机体造成损坏;而不可逆的氧化修饰则会导致细胞功能障碍和组织损伤,主要包括蛋白质赖氨酸的羰基化和酪氨酸的硝基化[3,4]. 蛋白质的羰基化是氧化应激最显著的生物标志物之一,它与众多疾病的病理状态和治疗相关[5,6]. 蛋白质羰基化的来源有多种途径,主要包括赖氨酸、精氨酸和脯氨酸的脱氨基,蛋白质和还原糖的非酶糖基化,多肽链的氧化裂解和蛋白质与脂质过氧化产物的结合[7]. 氧化修饰也会发生在细胞色素c(Cytochrome c,Cyt c)上,因其定位于线粒体内外膜之间,处在一个富含活性氧的环境中. 研究表明,Cyt c的氧化修饰起始于Tyr 67位残基的氧化,活性氧达到一定量时Met 80位残基发生亚砜化修饰和Lys 72/73位残基的羰基化,在氧化剂浓度更高的情况下则会发生更多残基的羰基化[8,9]. 蛋白侧链羰基化示意图如图1(a)所示(侧链氨基最终氧化成羰基). 蛋白质残基氧化形成活性羰基可以选择性地被吉拉德试剂(Girard’s Reagent T,GRT)标记,从而指示蛋白羰基化的程度[10-
图1
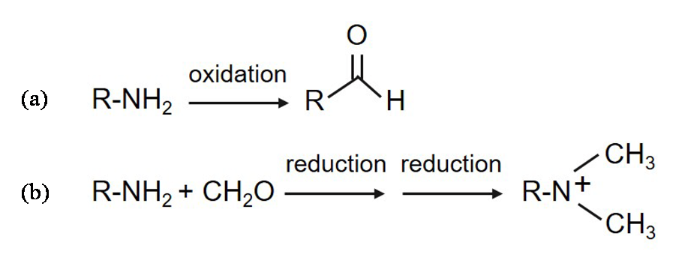
图1
蛋白侧链羰基化和还原甲基化示意图. (a)蛋白侧链羰基化;(b)蛋白侧链还原甲基化
Fig 1
Schematic diagram of protein side chain carbonylation and reductive methylation. (a) Protein side chain carbonylation; (b) Protein side chain reductive methylation
Cyt c在被各种氧化剂,例如过氧化氢(Hydrogen peroxide, H2O2)进行氧化修饰时会发生一定构象的改变[13,14],但目前尚不清楚蛋白的构象改变是否也会导致Cyt c发生氧化修饰. 本文主要研究蛋白构象转变对其氧化修饰的影响. 部分去折叠的Cyt c相较于自然状态的Cyt c结构更加松散,Cyt c与心磷脂(Cardiolipin, CL)相互作用之后其结构也会部分打开,且更接近原位条件. 据报道,蛋白质甲基化会改变其结构[15],因此我们还研究了Cyt c发生甲基化后,其氧化修饰程度所受到的影响. 为保护赖氨酸残基,所有的赖氨酸残基末端均被甲基化以防止羰基化,所选用方法为选择性还原甲基化,以二甲胺硼烷(Borane Dimethylamine Complex,DMAB)作为还原剂[16],其原理示意图如图1(b)所示. 基于此,我们研究了以下几种状态下人源Cyt c的氧化修饰,分别是处于自然状态下的Cyt c的氧化修饰、赖氨酸被保护的Cyt c的氧化修饰、部分去折叠状态下的Cyt c的氧化修饰和与CL相互作用下的Cyt c的氧化修饰.
1 实验部分
1.1 野生型人源Cyt c的表达和纯化
本文用到的试剂见表1.
表1 实验试剂
Table 1
| 试剂名称 | 纯度/浓度 | 生产厂家 | 规格 |
|---|---|---|---|
| 重水 | 氘代率99% | Sigma Aldrich | 100 mL |
| 胰蛋白胨 | AR | Oxiod | 500 g |
| 酵母提取物 | AR | Oxiod | 500 g |
| 氨苄青霉素 | AR | Biosharp | 20 g |
| 硫酸镁 | AR | 国药试剂 | 500 g |
| 葡萄糖 | AR | 国药试剂 | 500 g |
| σ-aminolevulinic acid | AR | 国药试剂 | 100 mg |
| 甘油 | AR | 国药试剂 | 500 mL |
| 氯化钙 | AR | 国药试剂 | 500 g |
| 氯化钠 | AR | 国药试剂 | 500 g |
| 氢氧化钠 | AR | 国药试剂 | 500 g |
| 盐酸 | AR | 国药试剂 | 500 mL |
| 氯化钾 | AR | 国药试剂 | 500 g |
| 磷酸二氢钠 | AR | 国药试剂 | 500 g |
| 磷酸氢二钠 | AR | 国药试剂 | 500 g |
| 磷酸二氢钾 | AR | 国药试剂 | 500 g |
| 硫酸亚铁 | AR | 国药试剂 | 500 g |
| EDTA-2Na | AR | 国药试剂 | 500 g |
| Tris | AR | 国药试剂 | 500 g |
| IPTG | AR | 国药试剂 | 25 g |
| 氯化锰 | AR | 国药试剂 | 500 g |
| 氯化钴 | AR | 国药试剂 | 100 g |
| 硫酸锌 | AR | 国药试剂 | 500 g |
| 氯化铜 | AR | 国药试剂 | 500 g |
| 硼酸 | AR | 国药试剂 | 500 g |
| 磷酸氢二钾 | AR | 国药试剂 | 500 g |
| 硫酸钠 | AR | 国药试剂 | 500 g |
| 溶菌酶 | AR | Sigma Aldrich | 10 g |
| 脱氧核糖核酸酶Ⅰ | AR | Sigma Aldrich | 1 mL |
| 盐酸胍 | AR | 国药试剂 | 500 g |
| 心磷脂 | 97 % | Sigma Aldrich | 100 mL |
| 吉拉德试剂T | 98 % | Sigma Aldrich | 100 g |
| DMAB | 97 % | Sigma Aldrich | 25 g |
| 甲醛水溶液 | 30% | 国药试剂 | 25 mL |
| 甘氨酸 | AR | 国药试剂 | 100 g |
野生型人源Cyt c表达和纯化操作如下:将重组人源Cyt c质粒导入到E.Coli BL21(DE3)感受态细胞中,涂于氨苄平板,倒置于培养箱中于37 ℃过夜培养;挑取单菌落至5 mL含有100 μg/mL氨苄的LB(Luria-Bertani)培养基中,在37 ℃,220 rpm培养箱中过夜培养,再将菌液转入1 L含有100 μg/mL氨苄的LB培养基中,于37 ℃,220 rpm培养箱中培养至OD600(600 nm的光密度值)>1,随后加入1 mL 1 000倍的IPTG(Isopropyl β-D-Thiogalactoside),在16 ℃,120 rpm条件下诱导约72 h后,在4 ℃,6 000 rpm条件下离心10 min收集菌体;将菌体沉淀重悬于含有5 mmol/L EDTA(Ethylene Diamine Tetraacetic Acid)的50 mmol/L Tris(Tris(hydroxymethyl)aminomethane)(pH 7.5)裂解液中,加入溶菌酶(1.5 mg/g湿菌体)和微量脱氧核糖核酸酶Ⅰ(DNase Ⅰ)消化1 h后,用高压破碎仪裂菌,将细胞裂解液在4 ℃、20 000 rpm条件下离心30 min收集上清液,在上清液中缓慢加入硫酸铵(150 g/L)在4 ℃盐析约3~12 h后,于4 ℃、20 000 rpm条件下离心30 min后收集上清液,将上清液置于20 mmol/L PB(Phosphate Buffer)(pH 7.0)缓冲液中于4 ℃下过夜透析. 将透析后的蛋白用0.22 μm超滤膜过滤后上样至用缓冲液A(20 mmol/L PB,pH 7.0)预平衡后的SP(Sepharose High Performance)阳离子柱中,随后用缓冲液B(20 mmol/L PB, 1 mol/L NaCl,pH 7.0)进行20%~70%线性梯度洗脱,将收集到的蛋白用截留分子量为10 kDa的蛋白浓缩管浓缩至体积小于5 mL,经0.22 μm超滤膜过滤后上样至用缓冲液C(20 mmol/L PB,250 mmol/L NaCl,pH 7.0)预平衡好的HiLoad 16/600 Superdex 75 pg 75柱,收集目标峰后用截留分子量为10 kDa的蛋白浓缩管进行浓缩,用去离子水洗涤多次,分装,冷冻干燥,保存于-20 ℃待用.
1.2 甲基化Cyt c的样品制备
甲基化Cyt c样品制备实验如下:配制1 mL浓度为1 mmol/L的蛋白,加入360 μL浓度为1 mol/L用20 mmol/L磷酸钠盐缓冲液(pH 7.0)配制的DMAB溶液,DMAB的浓度为蛋白中所含赖氨酸的20倍. 加入60 μL甲醛水溶液(含甲醛30 % v/v),使甲醛浓度为蛋白中赖氨酸残基的40倍,在4 ℃条件下震荡反应2 h,再加入上述相同量的DMAB溶液和甲醛溶液,同样条件下反应2 h,之后加入180 μL的DMAB溶液,在4 ℃条件下震荡过夜反应. 加入2倍甲醛摩尔浓度的甘氨酸进行猝灭处理,接着用截留分子量为10 kDa的超滤管超滤多次,除去体系中残留的DMAB溶液和甘氨酸,取少量体积的样品进行质谱实验,确保甲基化完全.
1.3 CL脂质体样品制备
CL脂质体通过旋蒸法进行制备[17],其主要实验步骤如下:先用注射器抽取5 mL的CL乙醇溶液注射到圆底烧瓶中,将水浴锅温度设置为45 ℃,打开冷凝循环系统,抽真空,待乙醇完全蒸发,直至烧瓶底部的液体成为一层薄膜,用新鲜配制的提前在冰箱中预冷的缓冲液重新溶解该薄膜,完全溶解后,在冰水浴中超声约15 min,即可得分散均匀的CL脂质体,使用时将其稀释到相应浓度即可.
1.4 Cyt c的GRT衍生化实验
不同方式的样品氧化修饰处理流程图如图2所示. 其中,自然状态下Cyt c的氧化修饰实验流程如图2(a)所示:将纯化后冻干的蛋白用20 mmol/L的磷酸钠盐溶液(pH 7.0)溶解,稀释至终浓度为10 μmol/L,体积约为10 mL,蛋白与H2O2的摩尔比分别1 : 0、1 : 5、1 : 25 和1 : 50,加入H2O2后在室温下静置30 min待其充分反应,使用截留分子量为10 kDa的蛋白浓缩管超滤4~5次将体系中未反应完全的H2O2去掉. 再加入用20 mmol/L磷酸钠盐配置的终浓度为100 mmol/L的GRT试剂,保持体系终体积为10 mL,在室温下反应3 h,使蛋白充分进行羰基化标记,使用截留分子量为10 kDa的蛋白浓缩管超滤换液8次,以除去体系中残留的GRT试剂. 图2(b~c)分别为甲基化的Cyt c和部分去折叠状态下Cyt c的衍生化实验流程. 图2(d)为Cyt c与CL作用后的衍生化实验:将与CL互作后的样品经氧化处理后,加入体积约20 mL的含有0.3 mol/L磷酸钠盐缓冲液(pH 7.0)处理约30 min,充分破坏蛋白和CL的相互作用,用截留分子量为30 kDa的超滤管超滤多次去掉反应体系中未反应完全的H2O2,后续处理方法与其他样品一致.
图2
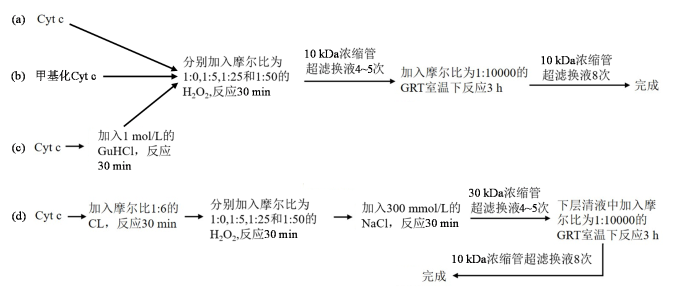
图2
Cyt c的GRT衍生化实验流程图
Fig 2
Schematic diagram of GRT derivatization experimental procedure for Cyt c
1.5 质谱实验
该实验通过Micro TOF-Q(Bruker)仪器采集数据,取150 μL甲基化的Cyt c放入质谱样品管中,放入样品池内,选择相应的质谱程序开始实验,实验耗时约20 min. 实验数据用Qualitative Analysis软件行处理.
1.6 核磁共振实验
所有的核磁共振(NMR)实验均在配备有TXI三共振超低温探头的Bruker Avance III 700 MHz的NMR谱仪上进行采集,实验温度为25 ℃. 一维氢谱的谱宽设置为20.00 ppm,谱中心为4.70 ppm,累加次数256;二维1H-13C HSQC实验采用HSQCETGP脉冲程序,1H和13C维的谱宽分别设置为20.00 ppm和60.00 ppm,谱中心分别为4.70 ppm和35.00 ppm,采样数据点阵t2×t1 = 2 048×128,累加次数64. 由于样品在处理过程中并没有加入内标,为了得到准确的分析数据,最终采用积分归一化的方法处理谱图. 调整每个谱图的基线使其保持一致,然后对每个谱相同化学位移范围内的GRT信号以及整个谱的所有信号分别进行积分,用GRT积分强度比所有信号积分强度,得到GRT信号积分占比.
1.7 圆二色谱实验
圆二色谱(Circular Dichroism,CD)实验在Chirascan圆二色谱仪上进行,实验温度为25 ℃,缓冲液体系为50 mmol/L磷酸钾溶液(pH 7.0),蛋白浓度为10 μmol/L,检测波长为180~260 nm,每个样品重复采样三次.
2 结果与讨论
2.1 GRT探针的NMR表征
在用GRT作为探针来表征Cyt c的氧化修饰之前,先用NMR方法对GRT进行表征,结果如图3所示. 在GRT的分子结构中,在其结构的末尾连接有三个甲基,这九个氢质子化学等价. 在1H NMR谱(图3(a))上,只看到两个明显的峰,因GRT的三甲基基团含有九个化学等价的质子,3.21 ppm处的信号峰来自该质子. 在1H-13C HSQC谱图中(图3(b)),在13C维化学位移约55 ppm处观察到一个峰,且1H维的化学位移与一维氢谱中的一致,因此可以判断这个峰即为GRT的信号峰. 该试剂中存在少量杂质,在一维氢谱中4.0 ppm附近和3.21 ppm信号峰左侧可以观测到强度较弱的峰,后者使得GRT质子信号在二维谱中不是完全对称. 无论在一维氢谱还是二维谱中,该信号均易于指认,因其不受邻近峰干扰、远离蛋白脂肪链甲基区,GRT探针非常适用于表征蛋白的氧化修饰.
图3
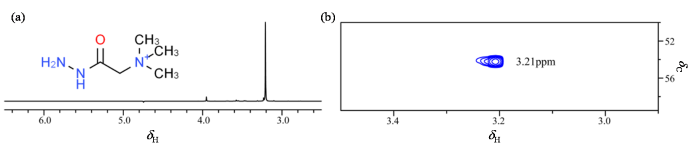
图3
GRT的分子结构图和NMR表征图. (a) GRT分子结构图和1H NMR谱图;(b) GRT试剂的1H-13C HSQC谱图
Fig 3
Molecular structure diagram and NMR characterization of GRT. (a) GRT molecular structure diagram and 1H NMR spectrum; (b) l 1H-13C HSQC spectrum of GRT reagent
2.2 甲基化Cyt c的氧化修饰结果分析
在人源Cyt c的序列中,共含有19个赖氨酸残基,当所有的赖氨酸都被甲基化后,理论上计算的分子量应该为12 762 Da(12 230+19×14×2=12 762 Da),质谱结果(图4(a))显示分子量为12 765 Da,与理论分子量接近,说明Cyt c通过还原甲基化的方法实现了所有赖氨酸残基的甲基化.
图4
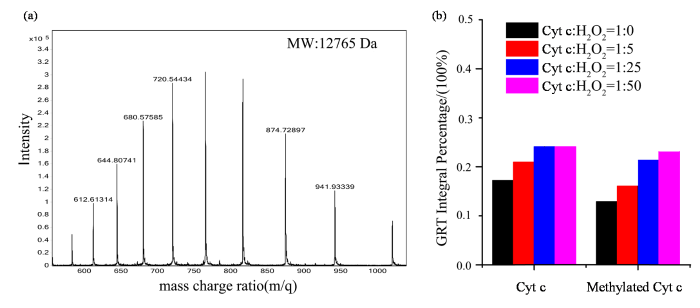
图4
(a) Cyt c还原甲基化后的质谱图;(b)甲基化Cyt c被H2O2氧化修饰的程度对比图
Fig 4
(a) Mass spectrum of reductively methylated Cyt c; (b) Comparative analysis of the extent of H2O2-induced oxidative modification of methylated Cyt c
在过量氧化剂的氧化修饰下,Cyt c中的残基很容易发生羰基化,通过甲基化保护赖氨酸残基时Cyt c的氧化修饰结果如图4(b)所示. 图4(b)中的第一组数据代表Cyt c在自然状态下时发生氧化修饰的结果,从数据中可以看出随着H2O2浓度的增加,GRT信号占比增加,表明蛋白的羰基化程度也逐渐增加,与此前报道一致. 第二组数据为Cyt c中的赖氨酸残基被甲基化保护起来后的氧化修饰结果,与自然状态下的氧化修饰的趋势一致,随着H2O2浓度的增加,GRT信号强度占比逐渐增强,蛋白羰基化的程度逐渐增加. 同时可以观察到在不同H2O2浓度的氧化修饰下,Cyt c的残基受保护时,其羰基化程度均小于对应的H2O2浓度下自然状态下的Cyt c的氧化修饰强度,这个结果表明通过甲基化保护赖氨酸的方法可以在一定程度上避免赖氨酸的羰基化,从而使蛋白整体羰基化程度降低.
2.3 部分去折叠状态的Cyt c的氧化修饰结果分析
Cyt c结构非常稳定,本研究中选择用1 mol/L的盐酸胍(GuHCl)来处理Cyt c,使其处于部分去折叠的状态[18],其CD结果如图5(a)所示. 在图5(b)中,第二组数据为部分去折叠状态的Cyt c在H2O2作用下发生的氧化修饰,可以看出经GuHCl处理后的蛋白在高浓度H2O2下的羰基化程度明显提高. 蛋白在部分去折叠后,结构变得更加松散,活性中心的血红素辅基会更加暴露,小分子更容易进入到蛋白的疏水口袋中,因此发生氧化修饰的程度更高. 该结果表明在氧化剂充足的情况下,蛋白内部更多的残基可以被羰基化. 至于低浓度H2O2下的羰基化程度有所下降,则可能与被GuHCl部分去折叠的蛋白区域对蛋白的整体活动性造成了一定的影响有关.
图5

图5
(a) Cyt c在1 mol/L GuHCl处理下的CD谱图;(b)部分去折叠Cyt c被H2O2氧化修饰的程度对比图
Fig 5
(a) Circular dichroism spectrum of Cyt c under 1 mol/L GuHCl treatment; (b) Comparative analysis of the extent of H2O2-induced oxidative modification of partially unfolded Cyt c
2.4 Cyt c在CL环境下的氧化修饰结果分析
以上两组实验观测到的都是溶液状态下Cyt c的结构变化对氧化修饰的影响,为进一步研究Cyt c在近原位条件下的结构变化对氧化修饰程度的影响,我们选择CL来模拟Cyt c在线粒体膜上的环境. 有研究显示,Cyt c在和CL发生相互作用后,活性中心的Met80和血红素铁离子的配位键断裂,蛋白的结构变得更加松散,导致过氧化物酶活性增高[19-
图6

图6
(a) CL的1H NMR谱图;(b-d) Cyt c与6倍摩尔比的CL相互作用后在不同H2O2浓度下的下层清液1H NMR谱图;(e)
Fig 6
(a) 1H NMR spectrum of CL; (b-d) 1H NMR spectra of the subnatant following the interaction of Cyt c with a 6-fold molar excess of cardiolipin and subsequent treatment with varying concentrations of H2O2; (e) 1H NMR spectra of the retentate and filtrate fractions obtained after ultrafiltration of the sample shown in
2.5 讨论
与赖氨酸残基受保护的Cyt c相比,部分去折叠的Cyt c在氧化剂的作用下可以增加其氧化修饰水平,在CL膜环境下时Cyt c与之进行相互作用后能则达到更高的氧化修饰水平. 通过对比Cyt c在不同结构状态下的氧化修饰程度,可以看到蛋白本身的结构特征与蛋白的氧化修饰程度有直接关系. 对Cyt c而言,整体结构的松散或者紧密状态,很大程度上决定了蛋白的氧化修饰程度. 文中的数据表明蛋白质的结构越松散,血红素辅基暴露的程度越高,其越容易发生氧化修饰,能达到的氧化修饰程度也越高. 但当Cyt c本身结构就相对松散时,在过高浓度的氧化剂条件下,氧化修饰过程中可能引发如蛋白聚集等其他过程,则可能反过来影响蛋白最终的氧化修饰水平.
3 结论
本文通过GRT试剂作为探针研究了不同结构特性Cyt c的结构和氧化修饰程度之间的关系. 发现蛋白质的结构越松散,越有利于发生氧化修饰. 最高的氧化修饰程度发生在Cyt c和CL相互作用后,说明CL对Cyt c的结构开放性有很大的促进作用. 但CL在打开Cyt c结构的同时会导致蛋白的聚集,反而对Cyt c的氧化修饰有抑制效果. 这说明在线粒体中,Cyt c结合在CL膜上可以加速Cyt c的氧化,但并不一定能使Cyt c被完全氧化. 另外,通过甲基化修饰蛋白中的赖氨酸残基可保护其不被氧化,从而降低了蛋白的羰基化程度.
利益冲突
无
参考文献
Free radical-mediated oxidation of free amino acids and amino acid residues in proteins
[J].
DOI:10.1007/s00726-003-0011-2
PMID:14661084
[本文引用: 1]

We summarize here results of studies designed to elucidate basic mechanisms of reactive oxygen (ROS)-mediated oxidation of proteins and free amino acids. These studies have shown that oxidation of proteins can lead to hydroxylation of aromatic groups and aliphatic amino acid side chains, nitration of aromatic amino acid residues, nitrosylation of sulfhydryl groups, sulfoxidation of methionine residues, chlorination of aromatic groups and primary amino groups, and to conversion of some amino acid residues to carbonyl derivatives. Oxidation can lead also to cleavage of the polypeptide chain and to formation of cross-linked protein aggregates. Furthermore, functional groups of proteins can react with oxidation products of polyunsaturated fatty acids and with carbohydrate derivatives (glycation/glycoxidation) to produce inactive derivatives. Highly specific methods have been developed for the detection and assay of the various kinds of protein modifications. Because the generation of carbonyl derivatives occurs by many different mechanisms, the level of carbonyl groups in proteins is widely used as a marker of oxidative protein damage. The level of oxidized proteins increases with aging and in a number of age-related diseases. However, the accumulation of oxidized protein is a complex function of the rates of ROS formation, antioxidant levels, and the ability to proteolytically eliminate oxidized forms of proteins. Thus, the accumulation of oxidized proteins is also dependent upon genetic factors and individual life styles. It is noteworthy that surface-exposed methionine and cysteine residues of proteins are particularly sensitive to oxidation by almost all forms of ROS; however, unlike other kinds of oxidation the oxidation of these sulfur-containing amino acid residues is reversible. It is thus evident that the cyclic oxidation and reduction of the sulfur-containing amino acids may serve as an important antioxidant mechanism, and also that these reversible oxidations may provide an important mechanism for the regulation of some enzyme functions.
Reversible and irreversible modifications of skeletal muscle proteins in a rat model of acute oxidative stress
[J].DOI:10.1016/j.bbadis.2009.09.011 URL [本文引用: 1]
Protein oxidation and cellular homeostasis: Emphasis on metabolism
[J].DOI:10.1016/j.bbamcr.2006.08.039 URL [本文引用: 1]
Metal-catalyzed oxidation induces carbonylation of peroxisomal proteins and loss of enzymatic activities
[J].
DOI:10.1016/j.abb.2005.04.018
PMID:15922287
[本文引用: 1]
Peroxisomes are involved in oxidative metabolic reactions and have the capacity to generate large amounts of reactive oxygen species that could damage biomolecules including their own resident proteins. The purpose of this study was to determine whether peroxisomal proteins are susceptible to oxidation and whether oxidative damage affects their enzymatic activity. Peroxisomal proteins were subjected to metal-catalyzed oxidation (MCO) with CuCl(2)/ascorbate and derivatized with 2,4-dinitrophenylhydrazine which allowed for spectrophotometric quantification of carbonylation. Immunochemical detection of carbonylated peroxisomal proteins, resolved by gel electrophoresis and detected with anti-DNP antibodies, revealed five oxidatively modified proteins with the following molecular weights: 80, 66, 62, 55, and 50 kDa. The proteins at 66, 62, and 55 kDa were identified as malate synthase (MS), isocitrate lyase, and catalase (CAT), respectively. MS and CAT were estimated to contain 2-3 mol of carbonyl/mol of protein as a result of MCO. Enzymatic assays revealed varying degrees of activity loss. Isocitrate lyase and malate synthase showed significant loss of activity while catalase and malate dehydrogenase were less inhibited by carbonylation. Our findings show that peroxisomal proteins are vulnerable to MCO damage and thus may also be affected by in vivo exposure to reactive oxygen species.
Is oxidative stress an issue in peritoneal dialysis
[J]?DOI:10.1111/sdi.v32.5 URL [本文引用: 1]
Protein carbonyl groups as biomarkers of oxidative stress
[J].DOI:10.1016/S0009-8981(03)00003-2 URL [本文引用: 1]
Protein carbonyls in meat systems: A review
[J].
DOI:10.1016/j.meatsci.2011.04.025
PMID:21621336
[本文引用: 1]
Protein oxidation (P-OX) is an innovative topic of increasing interest among meat researchers. Carbonylation is generally recognized as one of the most remarkable chemical modifications in oxidized proteins. In fact, the quantification of protein carbonyls by the dinitrophenylhydrazine (DNPH) method is the most common procedure for assessing P-OX in meat systems. Numerous studies have investigated the occurrence of protein carbonylation right after slaughter and during subsequent processing and cold storage of meat. However, the significance of protein carbonylation in meat systems is still poorly understood. Beyond their role as markers of protein oxidation, specific protein carbonyls such as α-aminoadipic and γ-glutamic semialdehydes (AAS and GGS, respectively) are active compounds that may be implicated in several chemical reactions with relevant consequences on meat quality. The formation of protein carbonyls from particular amino acid side chains contribute to impair the conformation of myofibrillar proteins leading to denaturation and loss of functionality. Recent studies also highlight the potential impact of specific protein carbonyls in particular meat quality traits such as water-holding capacity (WHC), texture, flavor and its nutritional value. As a truly emerging topic, the results from current studies provide grounds from the development of further investigations. The present paper reviews the current knowledge on the mechanisms and consequences of protein carbonylation in meat systems and aims to encourage meat researchers to accomplish further investigations on this fascinating research topic.Copyright © 2011 Elsevier Ltd. All rights reserved.
NMR study on the mechanism of cytochrome c methionine oxidation
[J].
细胞色素c甲硫氨酸氧化机制的NMR研究
[J].
DOI:10.11938/cjmr20222996
[本文引用: 1]
细胞在呼吸作用中产生活性氧,活性氧含量处于低水平时有利于信号传导,累积过量时会引发蛋白质氧化修饰.细胞色素c是位于线粒体内的多功能金属蛋白,其发生氧化修饰特别是甲硫氨酸Met80的亚砜化修饰可能影响蛋白构象变化,但其机制仍不清楚.本研究通过<sup>13</sup>C选择性标记细胞色素c上甲硫氨酸的末端甲基,利用液体核磁共振技术,追踪了细胞色素c在氧化环境中的氧化修饰变化.发现在氧化环境中,该蛋白质先由还原态转化为氧化态,当活性氧达到一定量时,再发生甲硫氨酸Met80的亚砜化修饰,但在活性氧作用初期未导致明显的蛋白结构变化.这表明细胞色素c具有一定抵御活性氧的作用.
Lysine carbonylation is a previously unrecognized contributor to peroxidase activation of cytochrome c by chloramine-T
[J].DOI:10.1039/C8SC03624A URL [本文引用: 1]
Characterization of oxidative carbonylation on recombinant monoclonal antibodies
[J].
DOI:10.1021/ac4039866
PMID:24731230
[本文引用: 1]
In the biotechnology industry, oxidative carbonylation as a post-translational modification of protein pharmaceuticals has not been studied in detail. Using Quality by Design (QbD) principles, understanding the impact of oxidative carbonylation on product quality of protein pharmaceuticals, particularly from a site-specific perspective, is critical. However, comprehensive identification of carbonylation sites has so far remained a very difficult analytical challenge for the industry. In this paper, we report for the first time the identification of specific carbonylation sites on recombinant monoclonal antibodies with a new analytical approach via derivatization with Girard's Reagent T (GRT) and subsequent peptide mapping with high-resolution mass spectrometry. Enhanced ionization efficiency and high quality MS(2) data resulted from GRT derivatization were observed as key benefits of this approach, which enabled direct identification of carbonylation sites without any fractionation or affinity enrichment steps. A simple data filtering process was also incorporated to significantly reduce false positive assignments. Sensitivity and efficiency of this approach were demonstrated by identification of carbonylation sites on both unstressed and oxidized antibody bulk drug substances. The applicability of this approach was further demonstrated by identification of 14 common carbonylation sites on three highly similar IgG1s. Our approach represents a significant improvement to the existing analytical methodologies and facilitates extended characterization of oxidative carbonylation on recombinant monoclonal antibodies and potentially other protein pharmaceuticals in the biotechnology industry.
Identification and quantification of protein carbonylation using light and heavy isotope labeled Girard's P reagent
[J].
PMID:16996067
Protein carbonyls are one of the most widely studied markers of oxidative stress. Determining increases in the concentration of protein carbonyls known to be associated with neurodegenerative diseases, heart disease, cancer and ageing. Identification of carbonylation sites in oxidized proteins has been a challenge. Even though recent advances in proteomics has facilitate the identification of carbonylation sites in oxidized proteins, confident identification remains a challenge due to the complicated nature of oxidative damage and the wide range of oxidative modifications. Here, we report the development of a multiplexing strategy that facilitates confident carbonylated peptide identification through a combination of heavy and light isotope coding and a multi-step filtering process. This procedure involves (1) labeling aliquots of oxidized proteins with heavy and light forms of Girard's reagent P (GPR) and combining them in a 1:1 ratio along with (2) LC/MS and MALDI-MS/MS analysis. The filtering process uses LC/MS and MALDI-MS/MS data to rule out false positives by rejecting peptide doublets that do not appear with the correct concentration ratio, retention time, tag number, or resolution. This strategy was used for the identification of heavily oxidized transferrin peptides and resulted in identification 13 distinct peptides. The competency of the method was validated in a complex mixture using oxidized transferrin in a yeast lysate as well as oxidized yeast. Twenty-five percent of the peptides identified in a pure oxidized sample of transferrin were successfully identified from the complex mixture. Analysis of yeast proteome stressed with hydrogen peroxide using this multiplexing strategy resulted in identification of 41 carbonylated peptides from 36 distinct proteins. Differential isotope coding of model peptides at different concentrations followed by mixing at different ratios was used to establish the linear dynamic range for quantification of carbonylated peptides using light and heavy forms of GPR.
Mass-spectrometry-based characterization of oxidations in proteins
[J].DOI:10.3109/10715762.2015.1023795 URL [本文引用: 1]
Insights on the conformational ensemble of Cyt c reveal a compact state during peroxidase activity
[J].
DOI:S0006-3495(19)30934-8
PMID:31810655
[本文引用: 1]
Cytochrome c (cyt c) is known for its role in the electron transport chain but transitions to a peroxidase-active state upon exposure to oxidative species. The peroxidase activity ultimately results in the release of cyt c into the cytosol for the engagement of apoptosis. The accumulation of oxidative modifications that accompany the onset of the peroxidase function are well-characterized. However, the concurrent structural and conformational transitions of cyt c remain undercharacterized. Fast photochemical oxidation of proteins (FPOP) coupled with mass spectrometry is a protein footprinting technique used to structurally characterize proteins. FPOP coupled with native ion mobility separation shows that exposure to HO results in the accumulation of a compact state of cyt c. Subsequent top-down fragmentation to localize FPOP modifications reveals changes in heme coordination between conformers. A time-resolved functional assay suggests that this compact conformer is peroxidase active. Altogether, combining FPOP, ion mobility separation, and top-down and bottom-up mass spectrometry allows us to discern individual conformations in solution and obtain a better understanding of the conformational ensemble and structural transitions of cyt c as it transitions from a respiratory role to a proapoptotic role.Copyright © 2019 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Cytochrome c as a peroxidase: activation of the precatalytic native state by H2O2-induced covalent modifications
[J].DOI:10.1021/jacs.7b07106 URL [本文引用: 1]
Structure and function of histone methylation binding proteins
[J].
DOI:10.1139/O08-129
URL
[本文引用: 1]
Chromatin structure is regulated by chromatin remodeling factors, histone exchange, linker histone association, and histone modification. Covalent modification of histones is an important factor in the regulation of the associated processes. The implementation and removal of various histone modifications have been implicated in DNA replication, repair, recombination, and transcription, and in RNA processing. In recent years, histone methylation has emerged as one of the key modifications regulating chromatin function. However, the mechanisms involved are complex and not well understood. A large volume of structural and biochemical information has been recently amassed for the Tudor, plant homeodomain (PHD), and malignant brain tumor (MBT) protein families. This review summarizes current knowledge of the structures and modes of recognition employed by the PHD, Tudor, and MBT domains in their interactions with target histone peptides.
Reductive methylation labeling, from quantitative to structural proteomics
[J].DOI:10.1016/j.trac.2019.07.009 URL [本文引用: 1]
pH dependence of ferricytochrome c conformational transitions during binding to cardiolipin membranes: Evidence for histidine as the distal ligand at neutral pH
[J].
DOI:10.1021/acs.jpclett.7b00597
PMID:28418677
[本文引用: 1]
The conformational changes of ferricytochrome c upon binding to cardiolipin-containing small unilamellar vesicles were studied at slightly acidic pH using fluorescence, visible circular dichroism, UV-visible absorption, and resonance Raman spectroscopy. The obtained spectroscopic response data suggest a mode of interaction, which is clearly distinct from the binding process observed at neutral pH. Evidence of a reversible and electrostatic binding mechanism under these conditions is provided through binding inhibition in the presence of 150 mM NaCl. Moreover, UV-visible absorption and resonance Raman spectra reveal that the conformational ensemble of membrane bound cytochrome c is dominated by a mixture of conformers with pentacoordinated and hexacoordinated high-spin heme irons, which contrast with the dominance of low-spin species at neutral pH. While our results confirm the L-site binding proposed by Kawai et al., they point to the protonation of a histidine ligand (H33) as conformational trigger.
Loss of either of the two heme-binding cysteines from a class I c-type cytochrome has a surprisingly small effect on physicochemical properties
[J].
DOI:10.1074/jbc.M004022200
PMID:10922364
[本文引用: 1]
Almost without exception, c-type cytochromes have heme covalently attached via two thioether linkages to the cysteine residues of a CXXCH motif. The reasons for the covalent attachment are not understood. Reported here is cytoplasmic expression in Escherichia coli of AXXCH and CXXAH variants of cytochrome c(552) from Hydrogenobacter thermophilus; remarkably, the single thioether bond proteins have, apart from an altered visible absorption spectrum, almost identical properties, including thermal stability and reduction potential, to the wild type CXXCH protein. In combination with previous work showing that an AXXAH variant of cytochrome c(552) is much less stable than the CXXCH form, it can be concluded that covalent attachment of heme via either of thioether bonds is sufficient to confer considerable stability and that these bonds contribute little to the setting of the reduction potential. The absence of AXXCH or CXXAH heme-binding motifs from bacterial cytochromes c may relate to the coexistence of the assembly pathway with that for formation of disulfide bonds in the bacterial periplasm.
Conformational properties of cardiolipin-bound cytochrome c
[J].
DOI:10.1073/pnas.1112312108
URL
[本文引用: 1]
\n Interactions of cytochrome\n c\n (cyt\n c\n ) with cardiolipin (CL) are important for both electron transfer and apoptotic functions of this protein. A sluggish peroxidase in its native state, when bound to CL, cyt\n c\n catalyzes CL peroxidation, which contributes to the protein apoptotic release. The heterogeneous CL-bound cyt\n c\n ensemble is difficult to characterize with traditional structural methods and ensemble-averaged probes. We have employed time-resolved FRET measurements to evaluate structural properties of the CL-bound protein in four dansyl (Dns)-labeled variants of horse heart cyt\n c\n. The Dns decay curves and extracted Dns-to-heme distance distributions\n P\n (\n r\n ) reveal a conformational diversity of the CL-bound cyt\n c\n ensemble with distinct populations of the polypeptide structures that vary in their degree of protein unfolding. A fraction of the ensemble is substantially unfolded, with Dns-to-heme distances resembling those in the guanidine hydrochloride-denatured state. These largely open cyt\n c\n structures likely dominate the peroxidase activity of the CL-bound cyt\n c\n ensemble. Site variations in\n P\n (\n r\n ) distributions uncover structural features of the CL-bound cyt\n c\n, rationalize previous findings, and implicate the prime role of electrostatic interactions, particularly with the protein\n C\n terminus, in the CL-induced unfolding.\n
Unravelling the non-native low-spin state of the cytochrome c-cardiolipin complex: evidence of the formation of a his-ligated species only
[J].
DOI:10.1021/acs.biochem.6b01281
PMID:28277678
The interaction between cytochrome c (Cyt c) and cardiolipin (CL) plays a vital role in the early stages of apoptosis. The binding of CL to Cyt c induces a considerable increase in its peroxidase activity that has been attributed to the partial unfolding of the protein, dissociation of the Met80 axial ligand, and formation of non-native conformers. Although the interaction between Cyt c and CL has been extensively studied, there is still no consensus regarding the conformational rearrangements of Cyt c that follow the protein-lipid interaction. To rationalize the different results and gain better insight into the Cyt c-CL interaction, we have studied the formation of the CL complex of the horse heart wild-type protein and selected mutants in which residues considered to play a key role in the interaction with CL (His26, His33, Lys72, Lys73, and Lys79) have been mutated. The analysis was conducted at both room temperature and low temperatures via ultraviolet-visible absorption, resonance Raman, and electron paramagnetic resonance spectroscopies. The trigger and the sequence of CL-induced structural variations are discussed in terms of disruption of the His26-Pro44 hydrogen bond. We unequivocally identify the sixth ligand in the partially unfolded, non-native low-spin state that Cyt c can adopt following the protein-lipid interaction, as a His ligation, ruling out the previously proposed involvement of a Lys residue or an OH ion.
Track the conformational change of unlabeled yeast cytochrome c in cell homogenate using NMR
[J].
基于磁共振的胞浆中无标记酵母细胞色素c构象变化追踪
[J].
DOI:10.11938/cjmr20222985
[本文引用: 1]
细胞色素c(cytochrome c,cyt c)是一个重要的多功能蛋白. 在线粒体中,它作为载体传递电子. 而在细胞质中,它可能会作为凋亡起始因子启动细胞凋亡程序. 复杂的细胞质环境是否会对其构象产生影响,以及产生怎样的影响,目前仍然没有得到确证. 本研究通过无标记的基于甲基的核磁共振(nuclear magnetic resonance,NMR)技术追踪了野生型酿酒酵母iso-1 cyt c在酵母细胞匀浆液中的构象变化. 发现cyt c在细胞匀浆中至少存在4种不同的氧化态构象和1种还原态构象. 而且随着时间的推进,不同构象之间发生转换. 结果表明cyt c的构象会随环境改变,这可能与抵抗氧化应激相关.
NMR reveals the conformational changes of cytochrome c upon interaction with cardiolipin
[J].
DOI:10.3390/life11101031
URL
[本文引用: 1]
Cellular protein homeostasis in the lungs is constantly disrupted by recurrent exposure to various external and internal stressors, which may cause considerable protein secretion pressure on the endoplasmic reticulum (ER), resulting in the survival and differentiation of these cell types to meet the increased functional demands. Cells are able to induce a highly conserved adaptive mechanism, known as the unfolded protein response (UPR), to manage such stresses. UPR dysregulation and ER stress are involved in numerous human illnesses, such as metabolic syndrome, fibrotic diseases, and neurodegeneration, and cancer. Therefore, effective and specific compounds targeting the UPR pathway are being considered as potential therapies. This review focuses on the impact of both external and internal stressors on the ER in idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD) and discusses the role of the UPR signaling pathway activation in the control of cellular damage and specifically highlights the potential involvement of non-coding RNAs in COPD. Summaries of pathogenic mechanisms associated with the ER stress/UPR axis contributing to IPF and COPD, and promising pharmacological intervention strategies, are also presented.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}